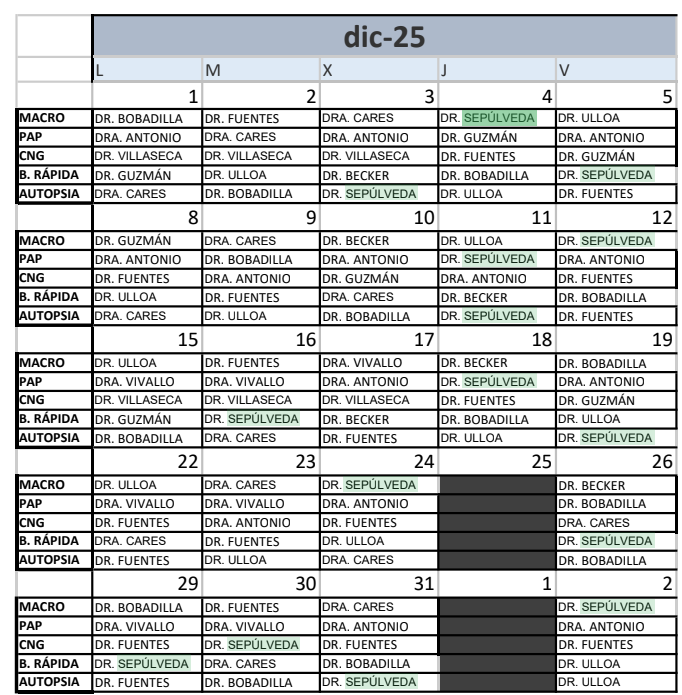
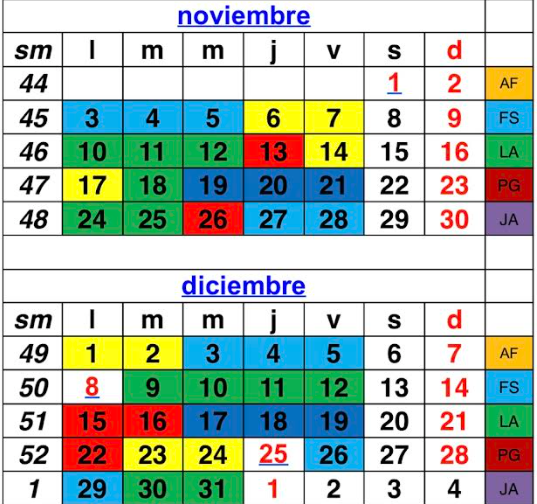
Calendarios
El Tiempo
Estructura y Duración de la Presentación
- Duración Total: La presentación tendrá una duración máxima de 20 minutos.
- Ronda de Preguntas: Después de la presentación, se destinarán 10 minutos para la interrogación de cada uno de los integrantes del grupo.
Metodología de la Presentación
- Selección Aleatoria: La exposición será iniciada por un integrante del grupo designado al azar por el docente.
- Interrupciones y Rotación: El profesor podrá interrumpir al expositor en cualquier momento para ceder la palabra a otro estudiante del mismo grupo.
- Objetivo de la Metodología: Este formato busca asegurar que todos los miembros del equipo estén preparados de manera uniforme y conozcan la totalidad del proyecto. El propósito es evitar presentaciones memorizadas que puedan ocultar la falta de participación o conocimiento de algunos integrantes.
Escala de Calificación y Conceptos Cualitativos
La calificación final se basará en una escala de 1 a 7, con los siguientes conceptos asociados:
| Nota | Concepto |
|---|---|
| 1 | MUY DEFICIENTE |
| 2 | DEFICIENTE |
| 3 | INSUFICIENTE |
| 4 | SUFICIENTE |
| 5 | BUENO |
| 6 | MUY BUENO |
| 7 | EXCELENTE |
Parámetros de Evaluación Referenciales
A continuación, se detallan los criterios que se utilizarán para la deliberación de la calificación, junto con el puntaje asignado a cada nivel de desempeño.
1. TRABAJO EN EQUIPO
Evalúa la colaboración y participación equitativa de los miembros del grupo.
- 4 PUNTOS: La exposición demuestra una excelente planificación y trabajo en equipo. Todos los integrantes han colaborado, exponen y participan activamente.
- 3 PUNTOS: Todos los miembros del equipo demuestran un conocimiento global de la presentación y todos exponen, aunque se observan algunas variaciones en el nivel de participación.
- 2 PUNTOS: La exposición muestra cierta planificación, y aunque todos participan, el nivel de intervención no es equitativo.
- 1 PUNTO: La presentación se percibe como individualista y sin colaboración evidente. No todos los miembros exponen al mismo nivel.
2. ORGANIZACIÓN DE LA INFORMACIÓN
Mide la estructura y claridad con la que se presenta el contenido.
- 4 PUNTOS: La información está muy bien organizada, presentada de forma clara y con una secuencia lógica.
- 3 PUNTOS: La mayor parte de la información está organizada de manera clara y lógica, aunque alguna diapositiva o sección puede estar fuera de lugar.
- 2 PUNTOS: No se aprecia un plan claro en la organización de la información, lo que genera cierta dispersión en el contenido.
- 1 PUNTO: La información se presenta de forma dispersa y desorganizada.
3. CONTENIDO
Se enfoca en el dominio y la precisión del tema expuesto.
- 4 PUNTOS: Los expositores demuestran un excelente dominio del tema, sin cometer errores ni mostrar dudas.
- 3 PUNTOS: Se demuestra un buen entendimiento de las partes más importantes del tema. La exposición es fluida y se cometen pocos errores.
- 2 PUNTOS: Los estudiantes necesitan hacer algunas rectificaciones durante la presentación y en ocasiones dudan al exponer.
- 1 PUNTO: Se realizan rectificaciones de manera continua. El contenido presentado es mínimo y no se demuestra un conocimiento adecuado del tema.
4. COMPRENSIÓN DEL TEMA
Evalúa la capacidad de los estudiantes para responder a las preguntas del docente.
- 4 PUNTOS: Responden con total precisión a todas las preguntas planteadas sobre el tema.
- 3 PUNTOS: Responden con precisión a la mayoría de las preguntas planteadas.
- 2 PUNTOS: Responden con precisión solo a algunas de las preguntas formuladas.
- 1 PUNTO: No logran contestar las preguntas planteadas por el docente.
TUMORES NEUROENDOCRINOS
Generalidades
- Los tumores neuroendocrinos bien diferenciados (WDNTs, tumores carcinoides) del intestino delgado constituyen aproximadamente un tercio de las neoplasias del intestino delgado.
- Parecen estar sobrerrepresentados en afroamericanos (38).
- Derivan de dos divisiones embrionarias separadas del tracto alimentario:
- Intestino anterior (foregut): Duodeno.
- Intestino medio (midgut): Yeyuno e íleon.
- Estos diferentes orígenes se correlacionan con una diferenciación neuroendocrina y un comportamiento clínico distintos.
Clasificación por Localización
1. Tumores Neuroendocrinos Bien Diferenciados (WDNTs) Duodenales
- Son el segundo tumor más común en el duodeno, después de los adenocarcinomas.
- Representan aproximadamente el 20% de los WDNTs del tracto gastrointestinal (39).
-
Epidemiología:
- Más comunes en hombres (ratio hombre-mujer de 1.5:1).
- Edad media de aparición: alrededor de los 60 años.
-
Tipos según Diferenciación Celular:
-
Diferenciación de células G (Gastrinomas):
- Componen dos tercios de los WDNTs duodenales (identificados por inmunotinción con anticuerpos contra gastrina).
- Se denominan gastrinomas (tumores de células G) si son funcionales y producen gastrina.
- Solo un tercio de los tumores con marcaje para gastrina son funcionales y causan el síndrome de Zollinger-Ellison (ZES).
- Estos tumores duodenales representan la mitad de los casos de ZES.
-
Expresión de Somatostatina (Somatostatinomas):
- Componen el tercio restante (tumores de células D).
- Se denominan somatostatinomas si producen hormona funcional.
- Un tercio de los tumores que expresan somatostatina están asociados con la enfermedad de von Recklinghausen (neurofibromatosis tipo 1/NF1).
- Los pacientes con NF1 a veces pueden tener la combinación de somatostatinomas, neurofibromas y tumores del estroma gastrointestinal (GISTs).
- El síndrome de somatostatinoma (diabetes mellitus, diarrea y colelitiasis) asociado a tumores de islotes pancreáticos casi nunca se observa en su totalidad con estos tumores intestinales.
- Tipo Indefinido: Un pequeño porcentaje.
-
Diferenciación de células G (Gastrinomas):
-
Características Clínicas y Localización:
- Los WDNTs del intestino delgado proximal casi nunca producen el síndrome carcinoide.
- Los gastrinomas y somatostatinomas funcionales se presentan en pacientes ligeramente más jóvenes y son algo más comunes en mujeres que los tumores no funcionales.
- Los tumores no funcionales que expresan gastrina suelen localizarse en el bulbo
duodenal.
- Un subgrupo se ha asociado con infección por Helicobacter pylori y uso prolongado de inhibidores de la bomba de protones. En este contexto, los tumores son pequeños (media 5.4 mm), se localizan en la mucosa y submucosa del bulbo, muestran un patrón insular en el examen histológico y tienen un comportamiento indolente. Algunos casos demuestran hiperplasia de células G en la mucosa no neoplásica (40).
- Los tumores funcionales se encuentran por igual en todas las partes del duodeno.
- Los somatostatinomas afectan típicamente la región periampular (41).
-
Características Macroscópicas:
- Típicamente son lesiones polipoides pequeñas, generalmente menores de 2 cm.
- Tienen base submucosa con una mucosa suprayacente que puede estar focalmente ulcerada.
- Los gastrinomas tienden a ser más pequeños que los somatostatinomas.
- Una minoría (15%) de los gastrinomas son multicéntricos.
- Pueden asociarse con MEN1 (neoplasia endocrina múltiple tipo 1).
- Pueden ser muy diminutos y aun así dar lugar a metástasis masivas en los ganglios linfáticos (44), lo que ha llevado a informes (inexactos) de “gastrinomas primarios de ganglios linfáticos”.
2. Tumores Neuroendocrinos Bien Diferenciados (WDNTs) del Intestino Delgado Distal
- Involucran principalmente el íleon, menos comúnmente el yeyuno (42), y raramente en asociación con divertículos de Meckel.
- Representan una quinta parte de los tumores neuroendocrinos del tracto GI.
-
Epidemiología:
- Edad media de diagnóstico: alrededor de los 60 años.
- Sin predilección por sexo.
- Tipo Celular: Tienden a ser WDNTs de células enterocromafines (células EC) que expresan serotonina.
-
Presentación Clínica:
- El síndrome carcinoide (diarrea acuosa, enrojecimiento y fibrosis endocárdica) se observa en el 5% al 7% de los casos.
- Los WDNTs yeyunoileales representan la mayoría de los casos de síndrome carcinoide en general.
-
Características Macroscópicas:
- Tienen el doble de probabilidades de ser multicéntricos (30%).
- Tienden a ser grandes (>2 cm) y a menudo están localmente avanzados en el momento de la presentación.
- Ocurren con afectación profunda de la pared intestinal y del mesenterio.
- Causan fibrosis mesentérica significativa, con la consiguiente obstrucción y acodamiento del intestino.
- También se puede observar isquemia intestinal franca debido a la angiopatía inducida por el tumor.
- Típicamente se centran en la mucosa profunda o submucosa, y la mucosa suprayacente está intacta o solo ligeramente erosionada.
3. Carcinomas Neuroendocrinos de Alto Grado (Carcinomas de Células Pequeñas)
- Se han reportado pocos casos primarios.
- Población: Hombres de 50 a 80 años.
- Localización: Típicamente en la región periampular.
- Pronóstico: Sombrío.
- Incidencia: Solo el 3.1% de los carcinomas de células pequeñas del tracto GI surgen en el intestino delgado; los sitios primarios más comunes son el colon (39%) y el esófago (30%) (43).
Diagnóstico
- Tumores Duodenales: Generalmente se diagnostican fácilmente mediante biopsia endoscópica.
-
Tumores Yeyunoileales:
- El diagnóstico es más problemático ya que a menudo no son accesibles por endoscopia/colonoscopia.
- Las técnicas de imagen convencionales (tomografía computarizada y estudios de contraste con bario) no son particularmente sensibles.
- Los análogos de somatostatina radiomarcados (octreotida y lanreotida) utilizados en estudios de gammagrafía han demostrado ser métodos de detección sensibles para lesiones primarias y metastásicas.
-
Errores Diagnósticos Potenciales (Pitfalls):
- En biopsias duodenales, las muestras aplastadas pueden simular focos de inflamación, tener una apariencia angiomatosa o de células fusiformes.
- Los tumores yeyunoileales pueden producir una respuesta fibroinflamatoria masiva que resulta en una gran masa mesentérica o retroperitoneal en los estudios de imagen. Las biopsias con aguja pueden mostrar características indistinguibles de la mesenteritis esclerosante o la enfermedad fibroesclerosante relacionada con IgG4. La inmunotinción para IgG4 a menudo muestra numerosas células plasmáticas marcadas, lo que confunde aún más el diagnóstico.
- Los paraganglios o paragangliomas pueden ser confundidos con neoplasias, especialmente en piezas de resección. La tinción con queratina ayuda en casos dudosos, ya que marca los WDNTs pero no los paraganglios.
Sistema de Gradación de la OMS
- G1 Menos de 2 mitosis/10 campos de alta potencia o un índice Ki-67 inferior al 2%.
- G2 Recuento mitótico de 2 a 20 por 10 campos de alta potencia o un índice Ki-67 del 3% al 20%.
- Grado 3 Carcinomas neuroendocrinos de alto grado (tipos de células grandes o pequeñas) caracterizados por más de 20 mitosis/10 campos de alta potencia o un índice Ki-67 superior al 20%.
-
Significado Pronóstico:
- La supervivencia es significativamente peor para los pacientes con tumores de grado 3 en comparación con los de grado 1 y 2.
- La supervivencia para los pacientes con tumores de grado 2 es peor que para aquellos con neoplasias de grado 1 (45).
- En las lesiones del intestino delgado, el marcaje con Ki-67 es un predictor pronóstico más preciso que la actividad mitótica.
Descripción Histopatológica
-
Características Generales:
- Proliferación monótona de células poligonales pequeñas y blandas con cantidades moderadas de citoplasma.
- Núcleos redondos y regulares con cromatina “en sal y pimienta”.
- El pleomorfismo y la actividad mitótica significativa son notablemente infrecuentes.
- Las características morfológicas de las células no difieren según la célula de origen.
- Generalmente son submucosos, no bien circunscritos, y las células tumorales frecuentemente se extienden en pequeños grupos hacia la mucosa y la pared intestinal.
-
Patrones Arquitectónicos:
- Variables y sin significado pronóstico, aunque pueden ser característicos de ciertos subtipos.
- Patrón Insular o Anidado (Tipo A): Visto en el carcinoma ileal.
- Patrón Trabecular (Tipo B).
- Patrón Acinar (Tipo C).
- A menudo, estos patrones se superponen y se observan múltiples patrones dentro de un mismo tumor.
-
Características Específicas del Subtipo:
- Tumores duodenales que expresan gastrina: Pueden manifestar cualquiera de los patrones arquitectónicos.
- Somatostatinomas: A menudo tienen características de un patrón de crecimiento acinar prominente con frecuentes cuerpos de psammoma intraluminales.
- WDNTs yeyunoileales: No muestran patrones de crecimiento característicos consistentes. Típicamente muestran un crecimiento anidado prominente, con más crecimiento trabecular y acinar en la periferia. Estos tumores suelen ser más grandes, se extienden profundamente en la pared intestinal y pueden asociarse con una fibrosis significativa.
- Carcinoma neuroendocrino de alto grado, tipo de células pequeñas: Típicamente muestran un artefacto de aplastamiento prominente y una alta actividad mitótica (>20 mitosis por 10 campos de alta potencia).
- Carcinoma ileal (tumor neuroendocrino bien diferenciado): Muestra nidos de células con núcleos que presentan un patrón de cromatina en “sal y pimienta” y formación prominente de rosetas. Los gránulos eosinofílicos prominentes son bastante característicos de los carcinoides ileales.
Inmunohistoquímica
- La mayoría de los WDNTs del intestino delgado son positivos para cromogranina y sinaptofisina, marcadores útiles para confirmar el diagnóstico.
- Aunque los WDNTs pueden mostrar evidencia inmunohistoquímica de producción de múltiples hormonas, esta información generalmente no es clínicamente significativa.
- Se pueden identificar tipos celulares específicos (expresores de gastrina, etc.) mediante marcaje inmunohistoquímico, pero su utilidad es limitada porque el estado funcional se correlaciona pobremente con la inmunohistoquímica.
- La tinción de queratina es positiva en los WDNTs, lo que ayuda a diferenciarlos de los paraganglios y paragangliomas (que son negativos).
Pronóstico
-
WDNTs Duodenales:
- Generalmente indolentes (mortalidad global de alrededor del 5%).
- Sin embargo, dos tercios de los somatostatinomas y la mitad de los gastrinomas funcionales se comportan de manera agresiva.
- El mal pronóstico se predice mejor por la invasión más allá de la submucosa y las metástasis ganglionares o a distancia.
-
WDNTs Yeyunoileales:
- Tienen un peor pronóstico que los del duodeno, con una tasa de mortalidad del 20%.
- La supervivencia se ha correlacionado negativamente con:
- Metástasis a distancia (hígado).
- Metástasis en ganglios linfáticos.
- Multiplicidad tumoral.
- Tasa mitótica / índice de marcaje Ki-67.
- Invasión más allá de la submucosa.
- Género femenino.
Asociación con Otras Neoplasias
Tanto los tumores neuroendocrinos de bajo como de alto grado en el intestino delgado a veces surgen en asociación con adenomas típicos.
Carcinoma de Células Renales de Células Claras
Definición / General
- El tumor epitelial renal más común, representando ~2% de todas las malignidades, típicamente con citoplasma claro y un patrón de crecimiento compacto anidado o acinar, intersectado por una vasculatura delicada y con alteraciones características en el cromosoma 3p que involucran la inactivación del gen VHL (von Hippel-Lindau).
Características Esenciales
- Masa cortical con superficie de corte variegada de color amarillo dorado, con arquitectura diversa, principalmente sólida y anidada.
- Citoplasma claro o eosinofílico granular y una red capilar prominente pero delicada.
- > 95% son esporádicos, mayormente una masa única, en la sexta a séptima década de vida, comúnmente albergando la inactivación del gen VHL localizado en el brazo corto del cromosoma 3 (3p25).
- Un pequeño porcentaje de tumores son familiares, principalmente en la enfermedad de VHL con tumores bilaterales múltiples y de aparición más temprana.
- Perfil inmunohistoquímico característico: positivo para PAX8, CAIX (tipo caja) y CD10; generalmente negativo para AMACR (35% positivo), CK7 (15% positivo) y CD117 (2% positivo).
Codificación ICD
- ICD-O: 8310/3 - Carcinoma de células renales de células claras.
Epidemiología
- ~2% de todas las malignidades.
- 65 - 70% de todos los carcinomas de células renales (RCC).
- La mayoría ocurre después de los 40 años, predominantemente en la sexta y séptima décadas.
- Ratio H:M = 1.5:1.
- Referencias: 30243799, 10768592, 30620402.
Localizaciones
- Riñón, típicamente como una masa cortical solitaria en tumores esporádicos.
- Múltiples tumores pueden representar síndromes familiares, pero la extensión venosa retrógrada desde el tumor esporádico dominante también es posible.
- La invasión del seno renal es la vía de diseminación más común, a menudo acompañada de invasión de la vena renal o sus ramas segmentarias, lo que lleva a un mayor riesgo de metástasis a distancia.
- Metástasis
- Hematógena es más común: pulmón (la más común), hueso, hígado, retroperitoneo, pleura, sistema nervioso central (SNC), cabeza y cuello (18076920, 31440453).
- Linfática es menos común en comparación con el RCC papilar y el RCC cromófobo: ganglios linfáticos hiliares, aórticos, cavos y torácicos (18782311, 18240184).
Fisiopatología
- Se sugiere que surge de las células epiteliales que recubren el túbulo contorneado proximal (37182269, 25387056).
- Pérdida de la función de la proteína VHL por un proceso de 2 pasos en 50 - 82% de los carcinomas de células renales de células claras somáticos (ccRCC), lo que lleva a la deleción, translocación no balanceada o alteración bialélica en el gen supresor de tumores VHL (von Hippel-Lindau) (3p25-26).
- El primer evento de pérdida de 3p, generalmente por cromotripsis, ocurre en la infancia o adolescencia en unas pocas cientos de células (29656891).
- El segundo alelo sufre una mutación somática o inactivación epigenética a través de la hipermetilación.
- La pérdida de la proteína VHL conduce a la acumulación del factor de transcripción inducible por hipoxia alfa (HIF1α).
- La acumulación de HIF1α impulsa la transcripción de genes asociados a la hipoxia, incluyendo VEGF, PDFGβ, GLUT1, TGFα, CAIX, EPO y metaloproteinasas (26838102).
- Otras mutaciones conductoras están presentes en porcentajes menores (ver Descripción Molecular / Citogenética).
Etiología
- Factores de riesgo: tabaquismo, obesidad, hipertensión, diálisis a largo plazo (particularmente en la enfermedad renal quística adquirida del adulto) e historia familiar de cáncer de riñón.
- Tumores familiares: principalmente la enfermedad de von Hippel-Lindau, menos comúnmente en familias que segregan translocaciones constitucionales del cromosoma 3, síndrome de predisposición a tumores BAP1, síndrome de Cowden, síndrome de Birt-Hogg-Dubé y esclerosis tuberosa (12872808, 26559379, 31326218, 23764071, 23752087).
Características Clínicas
- Más comúnmente: anemia, hematuria macroscópica, dolor en el flanco y masa.
- Pérdida de peso y fiebre en etapas tardías.
- La tríada clásica de masa en el flanco, dolor y hematuria está presente en < 10% de los casos (12474528).
Diagnóstico
- 60 - 80% se encuentran incidentalmente en imágenes radiológicas.
- Nefrectomía o nefrectomía parcial; el diagnóstico definitivo puede ser posible mediante biopsia con aguja.
Descripción Radiológica
- Tomografía computarizada (TC): exofítica con realce mixto y apariencia heterogénea (debido a necrosis interna, cambio quístico o hemorragia).
- La clasificación de Bosniak (categoría I a IV) para quistes renales guía el manejo aproximando el riesgo de malignidad.
- Resonancia magnética (RM): precisión similar a la TC, heterogénea en T1 e hiperintensa (brillante) en T2 cuando el uso de materiales de contraste está contraindicado.
- En casos metastásicos, la RM y la tomografía por emisión de positrones (PET / TC) son los métodos preferidos.
- Ecografía: útil para la detección incidental de masas renales.
- Referencia: 23382290.
Factores Pronósticos
- Supervivencia a 5 años: 50 - 70% después de la nefrectomía, 10% en enfermedad metastásica.
- La diferencia en la supervivencia se debe principalmente a diferencias en el estadio TNM y el grado nuclear, independientemente del tipo histológico del RCC (15837991, 15964127).
- Peor pronóstico dentro del mismo estadio: grado histológico más alto, diferenciación sarcomatoide y rabdoide y > 10% de necrosis tumoral coagulativa (33121821, 23348209, 25474516).
- Peor pronóstico que el RCC papilar y cromófobo.
- Mejor pronóstico con solo pérdida de VHL o mutación de PBRM1, mal pronóstico con múltiples mutaciones de genes conductores o pérdida de 4p, 9p o 14q.
- Los ccRCC con mutación en BAP1 tienen características morfológicas e inmunofenotípicas distintivas asociadas con un comportamiento más agresivo que los ccRCC típicos con mutación solo en VHL (33210135).
- La mutación en PBRM1 es mutuamente excluyente con la mutación en BAP1 y se asocia con una mejor respuesta a la inmunoterapia.
- Estadificación TNM: el predictor más preciso (ver Estadificación de
tumores
renales).
- pT1 y pT2: limitado al riñón, clasificado según el tamaño.
T1a(≤ 4 cm).T1b(> 4 cm a 7 cm).T2a(> 7 cm a 10 cm).T2b(> 10 cm).
- pT3: diseminación regional más allá del parénquima renal.
pT3a: diseminación extrarenal regional en la grasa perirrenal, grasa del seno renal, afectación de la vena renal o venas segmentarias o invasión del sistema pielocalicial.pT3b: extensión a la vena cava inferior por debajo del diafragma.pT3c: extensión del tumor a la vena cava inferior por encima del diafragma o invasión de la pared de la vena cava.
- pT4: diseminación a distancia, más allá de la fascia de Gerota, incluida la extensión contigua a la glándula suprarrenal ipsilateral.
- pT1 y pT2: limitado al riñón, clasificado según el tamaño.
- Gradación Histológica
- Sistema de gradación WHO / ISUP: 4 niveles, utiliza la prominencia nucleolar; se
usa
para carcinoma de células renales de células claras y papilar (el RCC cromófobo
no
se gradúa).
- G4: pleomorfismo nuclear extremo, células gigantes multinucleadas o diferenciación rabdoide o sarcomatoide.
- G3: nucléolos conspicuos y eosinofílicos a 100x de magnificación.
- G2: nucléolos conspicuos y eosinofílicos a 400x de magnificación (pero no prominentes a 100x de magnificación).
- G1: nucléolos ausentes o inconspicuos y basofílicos a 400x de magnificación (24661331).
- Sistema de gradación WHO / ISUP: 4 niveles, utiliza la prominencia nucleolar; se
usa
para carcinoma de células renales de células claras y papilar (el RCC cromófobo
no
se gradúa).
- > 50% de características eosinofílicas granulares pueden predecir una mala respuesta a la terapia con IL2 (16113605).
- Los RCC de células claras con > 40% de componente eosinofílico pueden tener un peor resultado clínico (Mod Pathol 2015;28:S217).
Reportes de Casos
- Hombres de 41, 55 y 68 años con metástasis de RCC de células claras que disminuyeron significativamente de tamaño o se resolvieron sin tratamiento quirúrgico u otros tipos de tratamiento (33250744).
- Hombre de 52 años con un RCC de células claras metastásico en el antebrazo sin una masa renal primaria identificable (31440453).
- Hombre de 63 años con un RCC de células claras de 15 cm con células gigantes sincitiales (15578891).
- Hombre de 65 años con un tumor de 2.4 cm con granulomas sarcoideos no necrotizantes (33063009).
Tratamiento
- La elección del tratamiento depende del estadio, la salud general del paciente y otros factores individualizados.
- La resección quirúrgica de los estadios 1 - 3 puede ser curativa, pero hasta el 33% recurrirá.
- Nefrectomía parcial para tumores más pequeños y localizados; nefrectomía radical para tumores de más de 4 cm y tumores localizados centralmente.
- Crioterapia y ablación por radiofrecuencia para tumores no resecables o algunos tumores pequeños.
- La quimioterapia sistémica tiene una eficacia limitada.
- La inmunoterapia con inhibidores de puntos de control tiene una tasa de respuesta del 15 - 20%: anticuerpos monoclonales contra PD-1 (nivolumab y pembrolizumab), PDL1 (avelumab y atezolizumab) y CTLA4 (ipilimumab) (31117033).
- Inhibidores de las vías de la diana de rapamicina en mamíferos (mTOR) (como temsirolimus).
- Inhibidores de la tirosina quinasa dirigidos a VEGFR (incluyendo lenvatinib, axitinib y sunitinib), PDGFR o multidirigidos (sorafenib, tivozanib, cabozantinib, lenvatinib y pazopanib) (26838102, 17045088).
- El inhibidor del factor inducible por hipoxia 2 alfa (HIF2α) belzutifan para ciertos tipos de tumores asociados a la enfermedad de VHL.
- Los inhibidores de puntos de control inmunitario han mostrado una respuesta significativa en ccRCC con características sarcomatoides y rabdoides.
- La combinación de inhibidores de la tirosina quinasa e inhibidores de puntos de control recibió la aprobación de la Administración de Alimentos y Medicamentos (FDA) para RCC avanzados (34991070).
- Terapia con IL2: se usa con menos frecuencia.
Descripción Macroscópica
- Típicamente una masa cortical renal unilateral y unicéntrica (tamaño promedio: 7 cm).
- Generalmente bien circunscrita por una pseudocápsula y un margen expansivo que protruye desde la corteza renal.
- Superficie variegada sólida y quística con áreas de fibrosis (gris) y hemorragia reciente o antigua (marrón); la necrosis y los cambios quísticos son comunes.
- Color amarillo dorado debido al alto contenido de lípidos.
- Los tumores de mayor grado pueden no ser amarillos debido a un menor contenido de lípidos y glucógeno.
- Áreas blandas y carnosas pueden reflejar diferenciación sarcomatoide.
- Afectación frecuente de la vena renal y el seno renal.
- Las masas bilaterales y multicéntricas son características de la enfermedad hereditaria.
- Notas sobre la estadificación del tumor
- Los tumores de más de 7 cm casi siempre invaden la grasa del seno renal; si no se observa invasión en tumores más grandes, se justifica una revisión macroscópica adicional (33121821).
- Invasión capsular
- Avance interrumpido e irregular del tumor hacia el tejido adiposo perirrenal y pérdida del contorno externo convexo y liso.
- Un tumor que protruye de forma lisa y está cubierto por la pseudocápsula del cáncer no se considera afectación de la grasa perirrenal.
- La célula tumoral debe tocar la grasa o extender lengüetas irregulares en los tejidos perirrenales, con o sin desmoplasia (28326247).
- Invasión del seno renal
- Ruta más común de diseminación extrarrenal.
- Suele ocurrir antes de la invasión capsular.
- A diferencia de la cápsula renal, el seno renal no está delimitado del parénquima renal por una cápsula fibrosa.
- No se considera invasión verdadera si el tumor está separado de las estructuras del seno por un borde de parénquima renal.
- Se considera invasión del seno si el tumor protruye en la grasa del seno renal claramente más allá del parénquima renal, incluso si está cubierto por tejido conectivo laxo (28326247).
- El tumor que rodea grandes estructuras linfovasculares es un signo de invasión de la grasa del seno (15577678, 10716160).
- Invasión vascular
- Puede mostrarse como nódulos tumorales en el seno renal.
- En vasos parcialmente obliterados, una sola capa de revestimiento endotelial sobre el tumor no excluye la invasión vascular.
- La afectación de venas pequeñas generalmente implica la afectación de venas grandes.
- La invasión del seno renal generalmente implica la invasión de la vena renal, se justifica un examen cauteloso (16145369).
Descripción de la Sección Congelada
- Aplicación limitada para determinar el subtipo de carcinoma de células renales.
- A veces es importante distinguir el carcinoma urotelial del RCC.
- Los hallazgos característicos en la microscopía incluyen citoplasma de células claras, vascularización muy densa (en malla de alambre) y patrón de crecimiento anidado.
- A veces solo están presentes núcleos anormales desnudos.
Descripción Microscópica (Histológica)
- Típicamente nidos compactos y láminas de células con citoplasma claro y membrana distinta.
- Citoplasma eosinofílico granular observado en tumores de alto grado o cerca de áreas de hemorragia o necrosis.
- Red de vasos pequeños y de paredes delgadas arborizantes (característica diagnóstica importante para casos con citoplasma eosinofílico granular).
- Patrones arquitectónicos: sólido, alveolar (anidado), acinar (tubular), microquístico (conteniendo glóbulos rojos extravasados o fluido eosinofílico) y ocasionalmente macroquístico.
- Puede observarse una arquitectura papilar focal, pero una formación papilar prominente plantea la posibilidad de otros subtipos (tumor renal papilar de células claras, RCC reordenado para TFE3, RCC alterado para TFEB, RCC con mutación en ELOC).
- Estroma: no descriptivo, sin reacción desmoplásica (a diferencia del carcinoma de conductos colectores o el carcinoma urotelial) con poca respuesta inflamatoria.
- Características de alto grado
- Diferenciación rabdoide: células malignas grandes de alto grado con abundante citoplasma eosinofílico homogéneo, núcleos excéntricos e inclusiones intracitoplasmáticas globulares eosinofílicas.
- Diferenciación sarcomatoide: puede ocurrir en cualquier subtipo de RCC (15087662).
- Necrosis tumoral.
- Variaciones histológicas poco comunes (significado pronóstico desconocido): quísticas, pseudopapilares, formación de hueso heterotópico, glóbulos hialinos intracelulares y extracelulares, inclusiones citoplasmáticas basofílicas, abundantes células gigantes multinucleadas, granulomas sarcoideos o características de myospherulosis (24499686, 37555895, 35347739).
- El ccRCC con mutación en BAP1 frecuentemente muestra arquitectura papilar, citoplasma eosinofílico y glóbulos citoplasmáticos (33210135).
- Prácticamente, las áreas de menor grado del tumor con histología clásica de ccRCC son las más útiles para el diagnóstico.
- Los tumores de mayor grado pueden demostrar características superpuestas con otros tipos de RCC.
Descripción Citológica
- Nidos cohesivos de células bastante uniformes con citoplasma pálido, mezclados con componentes estromales y capilares.
- Numerosas células individuales.
- Membranas celulares bien definidas, núcleos redondos centrales o excéntricos.
- Nucléolos prominentes en alto grado.
- Vacuolas intranucleares son comunes.
- Citoplasma pálido, vacuolado o granular.
- Fondo con sangre y necrosis es frecuente.
Tinciones Positivas (Inmunohistoquímica)
- Las tinciones positivas y negativas son menos fiables en tumores de mayor grado.
- PAX8: ~95%, nuclear; PAX2 similar pero menos sensible (19525927, 25025368).
- CAIX: marcador sustituto de alteraciones de VHL, 75 - 100%, membranoso difuso (tipo caja), bastante específico incluso en tumores con características sarcomatoides (25025368).
- CD10 (82 - 94%, membranoso) y RCC (72 - 84%, citoplasmático y membranoso): antígenos del borde en cepillo tubular proximal; también presentes en células normales.
- Marcadores epiteliales: AE1 / AE3, CAM 5.2, EMA / MUC1 (más sensible que AE1 / AE3).
- Vimentina (citoplasmática y membranosa).
Tinciones Negativas (Inmunohistoquímica)
- CK7: positiva hasta en un 15% de los casos; puede ser focalmente positiva o en parches en áreas de alto grado, componentes quísticos o células individuales en áreas mixoides y degenerativas del tumor.
- 34 beta E12.
- CK20 (25025368).
- AMACR: puede ser focalmente positivo en 25 - 50% de los casos (14707866, 32150988, 26457355).
- CD117.
- Cathepsin K.
- TFE3.
- HMB45.
- MelanA.
- Inhibina.
- La pérdida de BAP1 se correlaciona bien con la mutación del gen BAP1 (22683710).
Tabla Comparativa de Inmunohistoquímica
| PAX8 | CD10 | CAIX | RCC | Marcadores Melanocíticos | Vimentina | CK7 | HMWCK | CD117 / KIT | AMACR | GATA3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Clear cell RCC | + | + | + (tipo caja) | + | - | + | - | - | - | -/+ | - |
| Papillary RCC | + | + | +/- | + | - | + | + | - | - | + | - |
| Clear cell papillary RCT | + | - | + (tipo copa) | +/- | - | + | + | +/- | - | - | +/- |
| Chromophobe RCC | + | -/+ | - | +/- | - | - | + | - | + | - | +/- |
| Epithelioid angiomyolipoma | - | - | -/+ | - | + | -/+ | - | - | - | - | - |
| Tubulocystic carcinoma | + | + | +/- | + | - | +/- | -/+ | - | - | +/- | - |
| TFE3 rearranged / TFEB altered RCC | + | +/- | -/+ | +/- | TFEB: + TFE3: -/+ |
+ | - | - | - | +/- | - |
| MTSCC | + | -/+ | -/+ | +/- | - | + | + | -/+ | - | +/- | - |
| FH deficient RCC (FH loss and 2SC+) | + | - | - | - | + | - | - | - | - | -/+ | |
| TSC / mTOR / ELOC | + | + | + | - | -/+ | +/- | - | - | + | ||
| Leyenda | + = usualmente positivo | - = usualmente negativo | +/- = frecuentemente positivo | -/+ = ocasionalmente positivo |
Descripción en Microscopía Electrónica
- Gotículas de lípidos citoplasmáticos variables, escasos orgánulos, microvesículas y glucógeno.
- Evidencia de diferenciación tubular: microvellosidades largas y bien definidas, típicas del borde en cepillo visto en los túbulos proximales normales.
- Numerosas uniones celulares.
- Citoplasma granular eosinofílico debido a un mayor número de mitocondrias grandes y pleomórficas.
- Referencias: 3318057, 10976699.
Descripción Molecular / Citogenética
- Alteración del gen VHL en la mayoría de los casos somáticos.
- El locus 3p también contiene otros genes supresores de tumores remodeladores de cromatina frecuentemente asociados con ccRCC: PBRM1 (en 30 - 40% de los casos), BAP1 (6 - 10%) y SETD2 (7 - 11%) (23792563, 23620406, 23892664, 23797736).
- Otras alteraciones conductoras: genes de la vía mTOR (TSC1, TSC2, MTOR, PIK3CA y PTEN), TP53, pérdida de 4p, 9p o 14q, ocurriendo principalmente en asociación con alteraciones de VHL como conductores subclonales.
- La progresión del ccRCC conduce a un cariotipo poliploide y a más ganancias o pérdidas de material genético, contribuyendo a la progresión y agresividad de la enfermedad.
- Síndrome de cáncer familiar de Von Hippel-Lindau
- Alto riesgo de desarrollar tumores renales.
- Los pacientes nacen con un defecto de línea germinal en 1 de los 2 alelos del gen VHL.
- La pérdida del segundo alelo funcional (un "segundo golpe") conduce al desarrollo de la enfermedad clínica y la formación de tumores.
- Patrón de herencia autosómico dominante.
- Tiende a manifestarse a una edad temprana con un riesgo de por vida de desarrollar tumores renales de ~70%.
- ccRCC bilateral multifocal en el 35 - 45% de los pacientes.
- Se han reportado hasta 1,100 quistes y 600 focos microscópicos de ccRCC (14634372).
- Otros síndromes familiares (ver Etiología).
Ejemplo de Informe de Patología
- Riñón, biopsia por aguja gruesa:
- Carcinoma de células renales de células claras (ver comentario).
- Comentario: Las células tumorales muestran inmunotinción positiva para PAX8, CAIX y queratina CAM 5.2. CK7 es negativo, lo que argumenta en contra de un tumor renal papilar de células claras.
- Riñón, nefrectomía radical:
- Carcinoma de células renales de células claras, grado 3 de WHO / ISUP, midiendo 2.5 cm en su mayor dimensión (ver comentario).
- El tumor está confinado dentro de la cápsula renal.
- Todos los márgenes quirúrgicos son negativos para carcinoma.
- Ver informe de resumen de cáncer.
- Comentario: Las tinciones inmunohistoquímicas son positivas para PAX8, CAIX (patrón tipo caja) y CD10, lo que apoya este diagnóstico.
Diagnóstico Diferencial
- RCC Papilar con aclaramiento citoplasmático:
- Arquitectura papilar, carece de la prominente vasculatura delicada en "malla de alambre".
- Positivo para CK7 y AMACR; negativo para CAIX (patrón de tinción opuesto al RCC de células claras) (18779729).
- Tumor renal papilar de células claras (CCPRCT):
- Células tumorales de bajo grado con citoplasma claro (generalmente G1).
- Bien circunscrito, con arquitectura variable papilar, tubular, acinar y quística.
- Disposición nuclear luminal polarizada característica (en "teclas de piano") alejada de la membrana basal y positividad para CAIX tipo "copa", en lugar del patrón membranoso difuso tipo "caja" en el carcinoma de células renales de células claras.
- Carece de áreas de alto grado, necrosis tumoral e invasión local.
- La falta de vasculatura en "malla de alambre" y la presencia de nidos cerrados argumentan en contra de CCPRCT.
- Difusamente positivo para CK7 y negativo para CD10 (opuesto al RCC de células claras).
- La mayoría de los tumores se presentan como estadio pT1 con excelente resultado clínico.
- CCPRCT positivo para GATA3 (28705707).
- AMACR (opuesto a PRCC) es usualmente negativo.
- Carcinoma de células renales con deficiencia de fumarato hidratasa:
- Tumor de alto grado y agresivo, visto en edades más tempranas que el ccRCC.
- Nucléolos característicos tipo inclusión con aclaramiento perinucleolar.
- Positivo para IHC de 2SC (tanto nuclear como citoplasmático) y pérdida de la expresión de FH.
- Asociado con leiomiomas de la piel y el útero en casos de línea germinal (síndrome HLRCC).
- Negativo para CAIX.
- Neoplasia renal quística multilocular de bajo potencial maligno
(MCRNLMP):
- Células claras que recubren los delgados septos fibrosos de múltiples espacios quísticos o dentro de los septos fibrosos.
- Poco común, generalmente de menor grado asociado con un pronóstico excelente.
- Ausencia de nódulos sólidos expansivos.
- Patrón de tinción IHC similar al RCC de células claras, pero más comúnmente positivo para CK7 y menos a menudo positivo para CD10 (22982885).
- Angiomiolipoma epitelioide (AML):
- Vasos AML anormales característicos, músculo liso y lagos sanguíneos.
- Carece de los nidos característicos vistos en ccRCC.
- Puede parecerse a un ccRCC de alto grado.
- Buscar áreas de AML típico con grasa y vasos dismórficos.
- Positivo para HMB45, cathepsin K.
- Negativo para PAX8, CAIX, EMA y citoqueratinas (11145253).
- Carcinomas de células renales con reordenamiento de TFE3 y alteración
de
TFEB:
- Reordenamiento de TFE3 (cr Xp11) o alteración de TFEB (cr 6p21, reordenado o amplificado).
- Los RCC con reordenamiento de TFE3 y TFEB se ven en un rango de edad más joven (mediana: 31 años).
- Los RCC con amplificación de TFEB ocurren en pacientes mayores (mediana: 64 años) (12459622, 19228722, 24618616, 27565001).
- Núcleos de alto grado, citoplasma mixto claro y eosinofílico (no claro).
- Diferentes patrones arquitectónicos: tubulopapilar o trabecular, pero carece de los nidos cerrados clásicos del ccRCC.
- Positivo para TFE3 o TFEB, cathepsin K, CD10, AMACR y variable focalmente para CAIX, EMA y citoqueratinas (opuesto al RCC de células claras).
- RCC de células claras con septos fibromusculares prominentes y positividad para
CK7,
incluyendo el carcinoma de células renales con mutación en ELOC:
- Generalmente exhiben un comportamiento indolente.
- Citológicamente se asemejan a ccRCC con citología clara (principalmente tumores de bajo grado, excepto el RCC con mutación en ELOC).
- Arquitectónicamente, estroma fibromuscular prominente con gruesas bandas fibromusculares.
- Mutaciones en la vía mTOR o en los genes TSC1 / TSC2; pueden ser de línea germinal.
- No se observan alteraciones de VHL.
- El RCC con mutación en ELOC es mutuamente excluyente con VHL, PBRM1, TSC1, TSC2 o mTOR.
- Positivo para CK7 (fuerte), CD10 y CAIX membranoso completo (tipo caja).
- RCC Cromófobo:
- Masa circunscrita con una superficie de corte homogénea de color canela claro.
- Células grandes con bordes celulares duros y bien definidos, citoplasma finamente reticulado, borde nuclear irregular (raisinoide) y halo perinuclear.
- Carece de vasculatura en "malla de alambre".
- Positivo para CK7, E-cadherina, CD117; negativo para CAIX y vimentina (patrón de tinción opuesto al RCC de células claras) (25025368).
- Oncocitoma:
- Arquitectura anidada con estroma mixoide / hialinizado.
- Células con citoplasma eosinofílico a granular y núcleos pequeños y redondos.
- Carece de vasculatura en "malla de alambre".
- Positivo para CD117; negativo para vimentina y CD10 (patrón de tinción opuesto al RCC de células claras).
- Carcinoma corticosuprarrenal:
- Positivo para inhibina, MelanA y SF1.
- Negativo para PAX8 y EMA y queratinas.
- Carcinoma de células claras de ovario:
- Positivo para queratina 34 beta E12 y CK7.
- PAX8 negativo en sitios extrarrenales pero puede ser positivo en el riñón (29629948).
- Carcinoma de células claras de tiroides:
- Positivo para tiroglobulina y TTF1.
- PAX8 es positivo en ambos.
- Mesotelioma:
- Positivo para calretinina, mesotelina y CK5/6.
- Hemangioblastoma:
- Carece de atipia nuclear, nucléolos prominentes y necrosis.
- PAX8 negativo e inhibina positivo (opuesto al RCC de células claras).
- PAX8 negativo en sitios extrarrenales pero puede ser positivo en el riñón (29629948).
Estadificación del Carcinoma de Células Renales: Avances Actuales y Potenciales
Resumen
Las clasificaciones formales de estadificación para el carcinoma de células renales
(CCR) se
propusieron inicialmente en 1978 y se integraron en el sistema Tumor, Nódulos,
Metástasis
(TNM) de la Unión Internacional Contra el Cáncer (UICC). A lo largo de
seis ediciones,
los criterios de gradación han evolucionado, siendo la última
edición
publicada en 2016. Sorprendentemente, los criterios de la categoría T
no cambiaron de
2009 a 2016, aunque un erratum posterior incorporó modificaciones de
la octava edición de la Clasificación TNM
del Comité Americano
Conjunto sobre el Cáncer (AJCC).
Advertencia: Los tumores localizados se estadifican según el tamaño del
tumor primario. La
clasificación TNM reconoce que estos tumores pueden superar los
10 cm de diámetro,
lo cual es problemático, ya que hay evidencia de que casi todos los CCR de más
de
7 cm y una proporción sustancial de los de menos de 7 cm, muestran
diseminación
extrarrenal. La infiltración tumoral más allá de la cápsula renal en la grasa perirrenal
también se
categoriza como T3a, aunque su importancia clínica no está clara. La
clasificación de la
invasión microvascular dentro del seno renal y el significado pronóstico del tumor en vasos
pequeños dentro
del riñón requieren clarificación.
Introducción
La evaluación pronóstica del carcinoma se basa en dos características clave: la rapidez de proliferación del tumor y su extensión. Para muchos cánceres, la diseminación es el parámetro crítico, marcando la diferencia entre la posibilidad de curación y la paliación.
El concepto de que la diseminación tumoral es un factor pronóstico importante no es nuevo. En el
CCR, la presencia de metástasis se ha reconocido desde hace tiempo como un signo de
mal pronóstico.
Ya en 1858 se identificó la extensión venosa como un modo de diseminación, y en
1932
se formalizaron las clasificaciones de la extensión tumoral para cáncer de mama (Dukes) y
colorrectal.
No fue hasta 1958 que Flocks y Kadesky propusieron un sistema formal de
estadificación para el
CCR, dividiendo los tumores en cuatro grupos: intrarrenal, local, nodular y
diseminación
metastásica. Demostraron que un estadio creciente se asociaba con una menor probabilidad de
supervivencia a 5 y
10 años. Posteriormente, Petkovic (1959) dividió el estadio intrarrenal
1 de Flocks y
Kadesky en Estadios A y B, basándose en la presencia o ausencia de
penetración de la
(pseudo)cápsula tumoral. También fusionó todos los estadios con diseminación regional en el
Estadio
C, incluyendo, por primera vez, la invasión vascular como criterio de gradación.
Robson et al. (1963, 1969) propusieron otra clasificación de
estadificación para
CCR, destacando la importancia de la diseminación local del tumor:
- Estadio
1: Tumores intrarrenales. - Estadio
2: Extensión a la fascia de Gerota. - Estadio
3: Infiltración de la vena renal o ganglios regionales (3ay3baisladamente) o en combinación (3c).
El sistema de estadificación de Robson fue el estándar de oro hasta el establecimiento del
sistema de
estadificación TNM. La clasificación TNM, iniciada por Denoix en
1943, se
basa en la extensión anatómica del cáncer y es, para muchos tipos de malignidad, el parámetro
pronóstico más
potente. La primera clasificación tumoral se publicó en 1958 bajo los auspicios de
la
UICC, y la tercera edición de 1978 incluyó la estadificación para
CCR.
Evolución Histórica de la Clasificación TNM para CCR
Clasificación TNM 1978
La clasificación TNM de 1978 para CCR (Tabla 1) se asemejaba a los criterios de Petkovic,
definiendo los tumores
T1 y T2 como confinados al riñón.
T1: Tumores intrarrenales pequeños.T2: Tumores intrarrenales grandes.T3: Tumores con diseminación regional, subdivididos en:T3a: Invasión extrarrenal pero sin penetración de la fascia de Gerota; con subcategorías complejas:T3cap(invasión de la cápsula renal),T3pel(invasión de la pelvis renal),T3m(invasión microscópica de la vena renal).T3b: Invasión macroscópica de la vena renal.T3c: Infiltración de la vena renal y la vena cava infradiafragmática.
T4: Tumores con diseminación más allá de la anatomía regional del riñón:T4a: Invasión de estructuras adyacentes.T4b: Infiltración de la vena cava supradiafragmática.
No se mencionaba un punto de corte dimensional específico, dejando al clínico la decisión de qué constituía un tumor pequeño o grande.
Problemas y Dificultades: Los estudios de validación posteriores informaron
dificultades,
como la falta de un punto de corte de tamaño entre T1 y T2, y la
excesiva
complejidad de la clasificación T3. Se observó un solapamiento entre el tamaño
de los tumores
aparentemente intrarrenales y la evidencia microscópica de extensión extrarrenal.
Clasificación TNM 1987
La cuarta edición de la Clasificación TNM (Tabla
1) revisó el
sistema de estadificación para CCR:
T1: Tumores intrarrenales<2.5 cm.T2: Tumores intrarrenales>2.5 cm.T3: Simplificado, confinado a tumores que mostraban invasión directa de la glándula suprarrenal ipsilateral y tejidos perirrenales localizados.T3a: Incluye la invasión directa de la glándula suprarrenal y la extensión temprana a los tejidos perirrenales.T3b: Incorporó los anterioresT3b,T3cyT4bde1978.
T4: Simplificado, incluyendo todos los tumores que invadían más allá de la fascia de Gerota.
Consideraciones sobre el tamaño tumoral y pronóstico: La elección del punto
de corte de
2.5 cm era incierta. Aunque tumores <3 cm eran considerados
adenomas y rara vez
metastatizaban, se reconoce que CCR pequeños pueden metastatizar y mostrar
extensión
extrarrenal. El punto de corte de 2.5 cm se eligió porque tumores más pequeños
rara vez se
diseminan.
Infiltración de la glándula suprarrenal: Clasificada como T3a,
a pesar de su
rareza (0.8-2.5% de los casos) y su mal pronóstico (supervivencia a 5 años tan
baja como
20%).
Infiltración microscópica de la vena renal: Se omitió como criterio de estadificación; la invasión macroscópica de la vena renal se agrupó con la infiltración de la vena cava. La importancia pronóstica de la infiltración de la vena cava era debatida.
Infiltración más allá de la fascia de Gerota: Clasificada como T4, ya que usualmente
impedía la
curación quirúrgica.
Clasificación TNM 1997
El cambio principal en la clasificación TNM de 1997 (Tabla 1) fue el ajuste del punto de corte para el tamaño de
los tumores
intrarrenales:
T1: Tumores<7 cm.T2: Tumores>7 cm.
El punto de corte de 2.5 cm de 1987 era problemático debido a la
escasez de tumores
T1 y que la mayoría de tumores se clasificaban en la categoría de estadio superior.
Se propuso el
nuevo punto de corte de 7 cm para estratificación según el pronóstico. Sin embargo,
el debate sobre
el significado pronóstico del tamaño tumoral persistió.
Infiltración de la vena cava: Se dividió en invasión por debajo del diafragma (T3a)
y por encima del
diafragma (T3b).
Clasificación TNM 2002
La clasificación TNM de 2002 (Tabla
1) introdujo
cambios importantes en la comprensión del comportamiento del CCR, incorporando
observaciones sobre
la invasión del seno renal como principal vía de diseminación.
T1: Dividida en dos subcategorías, con un punto de corte de4 cm.T1a: Tumores intrarrenales<4 cm.T1b: Tumores intrarrenales>4 cmy<7 cm.
T2: Tumores intrarrenales>7 cm.T3a: Invasión extrarrenal localizada, incluyendo invasión directa de la glándula suprarrenal e infiltración en los tejidos perirrenales, incluida la grasa del seno renal.T3b: Infiltración de tumor macroscópicamente en la vena renal (incluyendo sus segmentos con músculo) o en la vena cava (o su pared) por debajo del diafragma.T3c: Infiltración de tumor en la pared de la vena cava por encima del diafragma.T4: Los criterios paraT4permanecieron sin cambios respecto a1997.
Clasificación TNM 2009
En la séptima edición de la clasificación TNM para CCR:
T1: La categoríaT1permaneció sin cambios (T1a <4cm,T1b >4cm-7cm).T2: Se extendió, con tumores divididos por un punto de corte de10 cm(T2a >7-10cm,T2b >10cm).T3a: Se clasificó la presencia de tumor en las ramas segmentarias (con músculo) de la vena renal, y la infiltración en la grasa perirrenal y/o del seno renal (peripélvica), no más allá de la fascia de Gerota.T3b: La presencia de tumor invadiendo la pared de la vena cava, aparentemente a cualquier nivel, se reclasificó comoT3c.T4: La infiltración directa de la glándula suprarrenal ipsilateral se trasladó deT3aT4(invasión más allá de la fascia de Gerota, glándula suprarrenal ipsilateral).
Clasificación TNM 2016
La octava edición de la clasificación TNM,
lanzada en
2016 para implementación en 2017:
- Categorías
T: Permanecieron inalteradas desde la séptima edición.T1a:<4 cm.T1b:>4 cmy≤7 cm.T2a:>7 cmy≤10 cm.T2b:>10 cm.T3a: Macroscópicamente en vena renal, ramas segmentarias (con músculo), grasa perirrenal y/o del seno renal (peripélvica), no más allá de la fascia de Gerota.T3b: Macroscópicamente en vena cava por debajo del diafragma.T3c: Macroscópicamente en vena cava por encima del diafragma, invade la pared de la vena cava.T4: Invade más allá de la fascia de Gerota, glándula suprarrenal ipsilateral.
- Categoría
N: La única modificación fue la eliminación de la categoría N2. Esto significa que N1 ahora representa metástasis en ganglios linfáticos, sin subdivisión adicional basada en el número. - Correcciones posteriores: Un erratum se publicó para alinear la clasificación de la
UICCcon laAJCC(ver siguiente sección).
Clasificación del Comité Americano Conjunto sobre el Cáncer (AJCC)
En 1987, se intentó unificar la clasificación TNM con la
AJCC, y ambas
fueron idénticas hasta 2016. En la octava
edición de la
clasificación AJCC:
T3a: La categoríaT3adifirió de laUICCal incluir la infiltración en el sistema pielocalicial.- Requisito de macroscopicidad: Se eliminó la necesidad de reconocer el tumor
macroscópicamente en la vena
renal para clasificarlo como
T3a. De manera similar, ya no era necesario identificar el tumor "macroscópicamente" en la vena cava para las categoríasT3byT3c. - Invasión venosa segmentaria: Ya no se requería que la invasión venosa segmentaria se limitara a vasos que contuvieran músculo.
Tras la publicación de la octava edición del manual
TNM de la
UICC, se publicó un erratum en línea que alineó la clasificación de la
UICC con la de
la AJCC. Estos cambios siguieron las recomendaciones de la Conferencia de Consenso
de la Sociedad
Internacional de Patología Urológica (ISUP) de
2012, que
abordó los factores pronósticos del CCR.
- Se acordó que la infiltración de la vena renal no siempre es visible macroscópicamente, por
lo que el tumor
identificado microscópicamente debe incluirse en la categoría
T3a. - En casos de duda, la presencia de tumor infiltrando la pared de la vena renal proporcionaba evidencia concluyente de diseminación tumoral y el tumor debía reclasificarse a un estadio superior.
- Se señaló que las venas segmentarias no siempre contienen músculo liso visible, por lo que no es necesario identificar músculo intramural para confirmar la diseminación venosa.
Factores Pronósticos Específicos
Tamaño del Tumor
Es bien reconocido que los CCR intrarrenales metastatizan con frecuencia, y la
consideración más
importante para estratificar estos tumores es distinguir aquellos verdaderamente intrarrenales y
curables
mediante nefrectomía, de aquellos con diseminación tumoral oculta.
- Recomendaciones de Bell: Históricamente, se consideraba que los
CCR <3 cmeran adenomas benignos y rara vez metastatizaban, pero esta noción ha sido descartada; losCCR <3 cmson tumores malignos. - Tasa de metástasis: Los tumores pequeños son poco propensos a desarrollar metástasis, con
una tasa del
3%reportada en tumores<3 cmen EE. UU. - Puntos de corte óptimos: Numerosos estudios han intentado determinar el punto de corte
óptimo para separar
tumores sin potencial metastásico de aquellos con probable recurrencia o metástasis,
proponiendo puntos de
corte de
4.0,4.5,5.0,5.5,6.5y7.0 cm. Aunque proporcionan información pronóstica, ninguno fue absoluto para separar todos los tumores intrarrenales de los que metastatizaron. - Limitaciones del tamaño como único predictor: El tamaño del tumor es una variable continua y cualquier punto de corte elegido mostrará diferencias significativas en el pronóstico entre los tumores por encima y por debajo de dicho punto.
- Evolución de puntos de corte en
TNM:1987:T1/T2dividido a2.5 cm(poco práctico, pocos tumoresT1).1997:T1/T2dividido a7 cm(excesiva corrección, la mayoría de tumores>7 cmy una parte sustancial de los<7 cmdesarrollan enfermedad metastásica).2009: Introducción de un punto de corte de10 cmpara subdividir la categoríaT2enT2ayT2b.- Retención en
2016: La subdivisión a10 cmse mantuvo en la edición de2016, a pesar de la evidencia de que la estratificación de tumores>7 cmno tenía significado pronóstico, lo cual se refuerza por la creciente comprensión de la relación entre el tamaño del tumor y la invasión del seno renal.
Invasión del Seno Renal y la Grasa Perirrenal
La identificación del seno renal como la principal vía de diseminación tumoral más allá del riñón
ha
proporcionado una comprensión crucial de la relación entre el tamaño del tumor y el pronóstico
en
CCR.
- Invasión temprana: En un estudio de
73 CCRde células claras, la invasión del seno renal se asoció significativamente con el tamaño tumoral. Aunque presente en tumores de1-2 cm, la incidencia aumentó con el tamaño (Tabla 2). - Estudios ampliados: La relación entre el tamaño tumoral y la invasión del seno renal se
mantuvo en una serie
de
120casos (Tabla 2). - Estudio reciente (
917 CCRde células claras):- En
178casos con tumores>7 cm, hubo invasión del seno renal en168casos (grasa del seno renal en9, invasión de la vena del seno renal en117, o combinación en42). - Solo
3 CCR >7 cmestaban confinados al órgano (tumores atípicos comoCCRquísticos o con infarto extenso). - La proporción de tumores con extensión extrarrenal (invasión del seno renal y/o infiltración de grasa perirrenal) aumentó con el tamaño tumoral (Tabla 2).
- En
- Invasión de la cápsula renal y seno renal: Bonsib sugirió que a medida que los tumores
crecen, eventualmente
alcanzan la cápsula renal fibrosa, que actúa como una obstrucción a la diseminación
extrarrenal. Sin
embargo, no hay impedimento para el crecimiento hacia la pelvis renal. La invasión del seno
renal es un
fenómeno relativamente temprano. Un tumor de
4-5 cmprobablemente abarca desde la cápsula renal hasta el seno renal, lo que implica una alta probabilidad de invasión del seno renal. - Pronóstico de la invasión del seno renal vs. grasa perirrenal: Un estudio de
205pacientes conCCRde células claras pT3a encontró que la invasión de la grasa perirrenal estaba en162(79%) y la invasión del seno renal en43(21%). La supervivencia específica por cáncer fue del41%y28%respectivamente, para estos dos grupos, indicando que la invasión del seno renal se asociaba con un riesgo significativamente mayor de muerte. Resultados contradictorios se presentaron en un estudio posterior del mismo grupo. - Problemas de muestreo: Los estudios anteriores a
2000, cuando se estableció la importancia pronóstica de la invasión del seno renal y el muestreo rutinario no era común, son limitados para el análisis de parámetros de estadificación. Un estudio encontró que en el42%de los casos deCCRde células claras pT1a que fallecieron por la enfermedad, había invasión de grasa del seno renal no reportada, y en el58%, invasión de vasos pequeños del seno renal.
Infiltración de la Vena Cava
La extensión tumoral a la vena cava, por encima y por debajo del diafragma, representa una enfermedad avanzada, aunque la intervención quirúrgica sigue siendo una posibilidad.
- Significado pronóstico: Estudios anteriores encontraron un significado pronóstico del nivel
de afectación de
la vena cava, pero estudios más recientes muestran resultados contradictorios. En una serie
grande de
CCR, se encontró una diferencia significativa en la supervivencia específica por cáncer entre pacientes con afectación de la vena renal y afectación de la vena cava. Sin embargo, no hubo diferencia en el pronóstico entre la invasión de la vena renal y la invasión de la vena cava inferior. - Clasificación clínica: Aunque el significado pronóstico del nivel de afectación de la vena
cava es
discutible, su ubicación tiene implicaciones quirúrgicas importantes. La clasificación
clínica es más
compleja que la del sistema
TNM, dividiéndose en cinco grupos:- Vena renal.
- Vena cava infrahepática.
- Vena cava intrahepática por debajo de los ostia de las venas hepáticas principales.
- Vena cava hepática que alcanza los ostia de las venas hepáticas principales.
- Vena cava suprahepática; trombo en la aurícula derecha.
Invasión Microvascular
El estado de la invasión microvascular como parámetro pronóstico para CCR es
incierto. La
clasificación TNM actual incluye la infiltración en las ramas segmentarias de la
vena renal como
característica de pT3, pero no menciona específicamente la
microvasculatura del seno renal o la infiltración de vasos, ya sea dentro del tumor o a
distancia.
- Estudios previos: Antes de la identificación del seno renal como principal portal de
diseminación, varios
estudios investigaron el significado pronóstico de la invasión microvascular, mostrando que
tenía
importancia pronóstica. La proporción de tumores con invasión microvascular varió
ampliamente (hasta
45%). - Limitaciones: La incidencia variable sugiere que muchos focos pequeños fueron pasados por alto, algunas series no fueron revisadas por expertos o no utilizaron marcadores inmunohistoquímicos.
- Extensión venosa retrógrada: Los tumores pueden mostrar extensión venosa retrógrada. Esto se
observa en los
riñones con tumor en el seno renal y la vena renal, posiblemente como resultado de la
obstrucción del flujo
venoso. Esta extensión puede llegar a la corteza renal, dando lugar a nódulos tumorales
separados de la masa
principal, lo que puede explicar la multinodularidad. Se observa en aproximadamente el
8%de losCCRde células claras y es más común en tumores grandes. - Red venosa corticomedular: La relación entre la invasión microvascular de la red venosa
corticomedular y el
pronóstico sigue siendo incierta, y la clasificación
TNMactual no incluye la invasión microvascular del seno renal ni del parénquima renal como criterio de estadificación.
Infiltración del Sistema Pielocalicial
La infiltración tumoral en el sistema pielocalicial se incluyó como característica de la
categoría de
estadificación T3a en la clasificación TNM
del
AJCC de 2017.
- Significado pronóstico: Si bien la afectación del sistema colector renal indica diseminación
tumoral
regional, su significado pronóstico sigue siendo incierto. La infiltración de la pelvis
renal se observa en
6.8-14%de losCCRy se ha asociado con un comportamiento tumoral agresivo en estudios previos. - Correlación con el pronóstico: Se ha correlacionado con el pronóstico en tumores localizados, aunque en algunos estudios no mostró un significado pronóstico independiente.
- Asociación con invasión microvascular: Un problema principal es que estos estudios incluyen
tumores grandes
(
T2) pero no informan sobre el estado del seno renal. Un estudio mostró una asociación significativa entre la invasión del sistema colector y la invasión microvascular. - Infiltración regional establecida: En tumores con afectación regional establecida, la presencia de afectación pielocalicial parece proporcionar información pronóstica, confiriendo un peor resultado para tumores con infiltración de estructuras regionales (pT3a).
Metástasis en Ganglios Linfáticos
Aunque la extensión de la vena renal es el modo más frecuente de diseminación del
CCR, las
metástasis también ocurren a través de los linfáticos hacia los ganglios hiliares.
- Clasificaciones tempranas: Las metástasis en ganglios linfáticos se incluyeron como característica de la diseminación tumoral regional.
- Visibilidad: Estudios previos han demostrado que los ganglios linfáticos con enfermedad
metastásica son
fácilmente visibles macroscópicamente en hasta el
80%de los casos. Sin embargo, se informa que la linfadenopatía se detecta quirúrgicamente en hasta el17%de los casos. - Categorización
TNM: Las clasificacionesTNMtempranas incluían una categorización compleja de tumores en ganglios linfáticos (Tabla 2). En la clasificación de1997, se simplificó a dos categorías según el número de ganglios positivos detectados. Las categoríasNde1997y2002no fueron respaldadas por estudios posteriores, ya que no hubo diferencia significativa en el pronóstico entre tumores pN1 y pN2. - Extensión extraganglionar: Se ha demostrado que la presencia de extensión extraganglionar
del tumor tiene
significado pronóstico. Estudios más contemporáneos indican que el número de ganglios
positivos no
proporciona información pronóstica adicional, lo que valida la categoría
Nactual de la clasificaciónTNM. - Diseminación linfática microvascular: Los estudios sobre la diseminación linfática
microvascular intrarrenal
de
CCRson limitados. Los linfáticos aumentan en número y tamaño desde la corteza media hasta la unión cortico-medular y, desde allí, hasta el seno renal. El tumor puede verse en los linfáticos adyacentes al frente del tumor, pero no parecen contribuir al desarrollo de la diseminación linfática. Sin embargo, la presencia de émbolos tumorales en los linfáticos preexistentes dentro del seno renal se asoció con metástasis en ganglios linfáticos, lo que apoyaría la inclusión del tumor dentro de los linfáticos del seno renal como una característica de la categoría T3a delTNM.
Conclusión
Importante: El sistema de estadificación TNM para el carcinoma
de células
renales es actualmente subóptimo. A pesar de la creciente evidencia de que los tumores
grandes se asocian
frecuentemente con la invasión del seno renal, la clasificación TNM ha
persistido en clasificar
tumores >7 cm y >10 cm como T2a y
T2b
respectivamente. Existe evidencia de que en estos tumores más grandes la diseminación
extrarrenal ya está
establecida. También es evidente que en tumores más pequeños puede demostrarse la invasión
del seno renal.
Esto significaría que todos los tumores clasificados como pT2, así
como un número significativo de tumores pT1b, están
subestadificados.
Por esta razón, se ha recomendado que las categorías de estadificación pT1-pT3a se revisen:
- pT1: Tumores
<4 cmde diámetro. - pT2: Tumores
>4-7 cm, considerando el potencial de invasión del seno renal. - Todos los tumores
>7 cmde diámetro deberían incluirse en la categoría de estadificación pT3a, ya que prácticamente todos presentan invasión del seno.
La definición actual de la invasión del seno renal es incierta y la presencia de invasión microvascular dentro del seno debería incluirse específicamente en los criterios de estadificación. Otras cuestiones a abordar incluyen el significado pronóstico relativo de la invasión microvascular intrarrenal, la extensión extracapsular y la infiltración tumoral en la grasa perirrenal, así como la estratificación de los tumores según el nivel de afectación de la vena cava.
Síndrome de Lynch (SL)
Definición
El Síndrome de Lynch (SL) es un trastorno autosómico dominante causado por mutaciones patogénicas
constitucionales en los genes de reparación de errores del ADN (MMR): MLH1,
MSH2,
MSH6 y PMS2.
Codificación
ICD-O:
609310Cáncer colorrectal, no polipósico hereditario, tipo 2120435Síndrome de Lynch I (cáncer colorrectal, no polipósico hereditario, tipo 1)614350Cáncer colorrectal, no polipósico hereditario, tipo 5614337Cáncer colorrectal, no polipósico hereditario, tipo 4613244Cáncer colorrectal, no polipósico hereditario, tipo 8
ICD-11:
Ninguno
Terminología Relacionada (No Recomendada):
"síndrome de cáncer familiar",
"cáncer colorrectal hereditario no polipósico".
Subtipos
Síndrome de Muir-Torre (MTS):
Co-ocurrencia de un tumor cutáneo sebáceo (adenoma sebáceo, sebaceoma, carcinoma sebáceo o queratoacantoma) con cualquier cáncer interno. Muchos pacientes con SL desarrollan tumores cutáneos sebáceos, por lo que pueden ser diagnosticados con MTS, pero no todos los pacientes con MTS tienen SL.
Síndrome de deficiencia constitucional de reparación de errores (CMMRD) y Síndrome de Turcot:
Condiciones alélicas debidas a mutaciones bialélicas en genes MMR. El CMMRD predispone a múltiples adenomas a una edad muy temprana, cáncer colorrectal, tumores cerebrales, leucemia, linfoma, características cutáneas similares a la neurofibromatosis tipo 1 y otras anomalías relacionadas con la deficiencia de reparación del ADN. La mayoría de los casos de síndrome de Turcot son variantes alélicas de CMMRD.
Localización
Los cánceres asociados al SL pueden surgir en el colon, recto, endometrio, estómago, intestino delgado, vesícula biliar, tracto hepatobiliar, páncreas, pelvis renal y/o uréter, vejiga, riñón, ovario, cerebro o próstata, dependiendo del gen afectado.
Características Clínicas
Predisposición a Cánceres:
El SL se caracteriza por una predisposición a una amplia variedad de cánceres, que pueden desarrollarse a cualquier edad, pero a menudo surgen en personas jóvenes.
Manifestación Variable:
Algunos individuos con SL desarrollan múltiples tumores, mientras que otros no desarrollan ninguno. La historia personal es importante, ya que la historia familiar por sí sola tiene un bajo valor predictivo.
Mutaciones de novo:
Se han descrito casos de SL debido a mutaciones germinales de novo.
Tipos de Cáncer Específicos:
Incluyen tumores de colon y recto, endometrio, estómago, intestino delgado, ovario, vesícula biliar, tracto hepatobiliar, páncreas, tracto urinario (pelvis renal, uréter y vejiga), riñón, cerebro y próstata, así como tumores cutáneos sebáceos.
Factores de Riesgo:
El riesgo de cáncer para un individuo con SL se ve afectado por el sexo, la edad, el gen afectado y el historial de cáncer.
Genes y Riesgo:
El riesgo de cáncer es mayor con mutaciones en MSH2 y MLH1, algo menor
(y de aparición
más tardía) cuando MSH6 está afectado, y aún menor con mutaciones en
PMS2.
Adenomas Colorrectales:
Los individuos con SL no suelen desarrollar un gran número de adenomas colorrectales, a menos que tengan otra condición predisponente.
Antecedentes Históricos:
- Descrito por primera vez por Warthin, luego redescripto por Lynch como
"síndrome de cáncer familiar". - Criterios de Ámsterdam (CA): Desarrollados para identificar familias aptas para la investigación genética, tienen baja sensibilidad y alta especificidad.
- Guías de Bethesda: Desarrolladas para seleccionar tumores para pruebas de SL, también sufren de baja sensibilidad.
"Cáncer colorrectal hereditario no polipósico"(DEPRECATED HNPCC): Término originalmente acuñado para la educación clínica, ya no se recomienda debido a su imprecisión y confusión.- Definición Actual: El SL se define por una mutación patogénica constitucional en los genes de reparación de errores de ADN.
Epidemiología
Prevalencia en Cáncer Colorrectal (CRC):
Aproximadamente 1 de cada 30 casos de CRC se debe al SL.
Prevalencia en la Población General:
Se estima en aproximadamente 1 de cada 125 (rango de 1 en 100-180), basándose en la penetrancia del SL como CRC a los 85 años (0.25) y los riesgos de CRC específicos de cada país.
Mutaciones Fundadoras:
Se han encontrado mutaciones fundadoras del SL en muchas poblaciones (ej. Finlandia), lo que resulta en una mayor prevalencia local.
Etiología
Causa Principal:
Mutación patogénica constitucional en un gen de reparación de errores de ADN (MLH1,
MSH2, MSH6 o PMS2).
Mutaciones en Genes Adyacentes:
Algunas personas con SL tienen mutaciones que involucran genes adyacentes y afectan o se
extienden a un gen MMR
(ej. EPCAM afectando a MSH2 o LRRFIP2 afectando a
MLH1).
Mecanismos Epigenéticos:
Algunos individuos pueden presentar mutaciones por mecanismos epigenéticos (metilación del ADN)
que afectan a
MLH1 o MSH2, algunas de las cuales pueden ser causadas por
reordenamientos que
involucran genes adyacentes.
Base de Datos InSiGHT:
La Sociedad Internacional de Tumores Hereditarios Gastrointestinales (InSiGHT) mantiene una base de datos pública de genes MMR y clasifica variantes genéticas.
Factores Modificadores:
- Aumento del riesgo: Tabaquismo, aumento del índice de masa corporal, consumo de alcohol.
- Reducción del riesgo: Ácido acetilsalicílico, ibuprofeno, suplementos multivitamínicos y de calcio, terapia de reemplazo hormonal (pero no anticonceptivos orales), y mayor paridad en mujeres.
Patogénesis
Inactivación Bialélica:
Las células no pierden la función de reparación de errores hasta que ambos alelos de un gen MMR dado son inactivados. Una "segunda mutación" somática en el alelo MMR normal convierte a la célula en deficiente en la reparación de errores (dMMR).
Consecuencias de la Deficiencia dMMR:
- Ventaja de Crecimiento: Las células dMMR escapan al control normal de la apoptosis y obtienen una ventaja de crecimiento relativa.
- Inestabilidad de Microsatélites (MSI): La deficiencia de reparación de errores aumenta la tasa de mutación puntual, especialmente en secuencias repetitivas de ADN llamadas microsatélites, manifestándose como MSI.
- Expresión Anormal de Proteínas MMR: La deficiencia puede llevar a una expresión anormal de proteínas MMR, detectable por inmunohistoquímica.
Deficiencia dMMR en Mucosa Colónica Normal:
Presente en aproximadamente un cripta por cm² (aproximadamente 10,000 criptas por individuo).
Vías hacia el Cáncer Colorrectal (CRC) en SL:
- Lesiones Planas con Mutaciones en
CTNNB1: Criptas dMMR pueden dar lugar a lesiones planas con mutaciones enCTNNB1(activando la víaWNT) en lugar deAPC, que se desarrollan en cánceres planos. Se cree que estas lesiones explican los cánceres de intervalo entre colonoscopias normales. - Progresión a Lesiones Polipoides: Estas lesiones planas pueden adquirir mutaciones
secundarias en
APCy volverse adenomatosas y polipoides. - Adenomas con Mutaciones Primarias en
APC: Los pacientes con SL pueden desarrollar adenomas con mutaciones primarias enAPC(como en la población normal), que adquieren secundariamente deficiencia de reparación de errores durante la progresión.
Nota: Los cánceres rectales con MSI suelen deberse a SL, aunque
no alberguen
mutaciones en CTNNB1, lo que sugiere que esta vía solo ocurre en el colon.
Deficiencia dMMR en Cánceres de Colon no-SL:
- Aproximadamente el 15% de los cánceres de colon no relacionados con SL tienen deficiencia de reparación de errores.
- La mayoría se deben a la hipermetilación bialélica somática esporádica del promotor del gen
MLH1, como parte de la vía de la lesión serrada sésil del lado derecho. - Estos tumores suelen (en ~85% de los casos) adquirir mutaciones específicas del oncogén
BRAF(mutacionesBRAF p.V600E), que pueden usarse para distinguirlos de los CRC no esporádicos. - Ocasionalmente, el CRC en pacientes con SL puede surgir a lo largo de esta otra vía.
- En algunos casos de CRC esporádico, la deficiencia de reparación de errores se debe a dos
mutaciones
somáticas en genes MMR, y puede estar asociada a otras condiciones hereditarias que afectan
la reparación
del ADN (ej. poliposis asociada a
MUTYHo poliposis asociada a errores de corrección de la polimerasa (PPAP)). Estos se describen como"síndrome de Lynch-símil". - Hipermetilación Constitucional del Promotor de
MLH1: Puede causar SL, generalmente esporádica, pero algunos casos son heredables debido a reordenamientos cromosómicos que causan metilación del promotor deMLH1. - Mutaciones Recesivas en
MSH3: Recientemente identificadas como causa de poliposis adenomatosa. - SL Digénico: Algunos individuos presentan mutaciones en más de un gen MMR, aunque no está claro si esto resulta en una enfermedad más grave que el SL causado por una sola mutación MMR.
Aspecto Macroscópico
La apariencia macroscópica está relacionada con el tipo de tumor y no es distintiva.
Histopatología
Características Típicas en CRC con MSI:
- Presencia de linfocitos infiltrantes del tumor.
- Reacción linfocítica peritumoral tipo Crohn.
- Pobre diferenciación.
- Características mucinosas y de células en anillo de sello.
- Patrón de crecimiento medular.
Estas características se observan tanto en cánceres esporádicos con MSI como en los que ocurren en el contexto de SL, pero no son lo suficientemente específicas para distinguir por sí solas los casos microsatélite-estables de los inestables.
Cánceres No Colorrectales Asociados a SL:
- Carcinomas gástricos: La mayoría son de tipo intestinal, <13% difusos y los mucinosos son muy raros. No se describe la presencia de linfocitos intraepiteliales.
- Carcinomas de intestino delgado: Frecuentemente muestran diferenciación mucinosa, de células en anillo de sello o medular, a menudo con linfocitos infiltrantes del tumor y reacción tipo Crohn, similar a los carcinomas ampulares.
- Otros carcinomas biliares: No tienen características distintivas.
- Cánceres pancreáticos: fuertemente asociados con SL son los carcinomas de células acinares y los carcinomas medulares.
Pruebas de Deficiencia de Reparación de Errores:
Las características histológicas de los cánceres asociados a SL no son específicas. Se recomienda
el cribado de
pacientes con CRC y/o otros tipos de cáncer, basado en la presencia de MSI o la ausencia de
proteínas MMR en el
tumor. No hay consenso sobre si la inmunohistoquímica o las pruebas moleculares son la primera
opción, y pueden
usarse en combinación. La inmunohistoquímica es preferible en casos con bajo porcentaje de
células tumorales o
intensa reacción inflamatoria. El análisis de mutaciones BRAF es una alternativa a
la prueba de
hipermetilación de MLH1.
Inmunohistoquímica
Uso:
Un primer paso común para el cribado de CRC para deficiencia de reparación de errores. Evalúa las
proteínas MMR
(MLH1, PMS2, MSH2 y MSH6).
Interpretación:
- Presencia de las cuatro proteínas: Sugiere estabilidad de microsatélites.
- Pérdida de tinción nuclear de cualquier proteína: Indica MSI y sugiere el gen probablemente involucrado, requiriendo pruebas adicionales.
- Pérdida de
MSH2sola o deMSH2yMSH6: Sugiere mutación enMSH2. - Pérdida de
MLH1sola o deMLH1yPMS2: Sugiere mutación subyacente o metilación enMLH1.
La pérdida concomitante de MSH2 y MSH6 (o de MLH1 y
PMS2)
refleja la unión heterodimérica de estas proteínas en los complejos de reparación.
Patrón de Expresión Típico:
Tinción difusa en los núcleos tumorales y en muchas células benignas (epiteliales, estromales, linfocitos).
Consideraciones en la Interpretación:
- Siempre debe realizarse con una tinción de control interno adecuada.
- Resultados Atípicos/Falsos Negativos:
- Tinción parcheada: Puede ocurrir por difusión desigual del anticuerpo, fijación variable o hipoxia tisular.
- Tinción citoplasmática: Se considera anormal/deficiente si no hay tinción nuclear.
- Expresión débil, parcheada, nucleolar o ausente de
MSH6: Reportada en tumores rectales post-tratamiento neoadyuvante sin MSI o mutación confirmada molecularmente. - Tinción heterogénea o pérdida de
MSH6: Ocasionalmente debido a una mutación secundaria (no germinal) en el tracto de mononucleótidos codificante deMSH6.
- SL con dMMR sin anomalías en IHQ: Aproximadamente 3-10% de los tumores con SL que tienen deficiencia de reparación de errores y MSI no muestran anomalías en inmunohistoquímica (presumiblemente por mutaciones que inhabilitan la función proteica pero la dejan detectable por IHQ).
Citología
No clínicamente relevante.
Patología Molecular Diagnóstica
Base Genética:
El SL se debe a la herencia autosómica dominante de una mutación constitucional que afecta uno de
los genes MMR:
MSH2 (2p21), MLH1 (3p22.2), MSH6 (2p16.3) y
PMS2 (7p22.1).
Base de Datos InSiGHT:
Recopila y clasifica variantes de genes MMR.
Deleciones de EPCAM:
- Extremo 3' sin extensión a
MSH2: Riesgo de cánceres gastrointestinales, pero no de cánceres no gastrointestinales característicos del SL. - Extensión a
MSH2: Fenotipo indistinguible de pacientes con mutaciones enMSH2.
Detección de Inestabilidad de Microsatélites (MSI):
La reparación defectuosa de errores impide el reconocimiento y la reparación de inserciones o
deleciones que
ocurren durante la replicación del ADN en secuencias repetitivas. MSI se define como la
presencia de alelos
extra en un microsatélite comparado con el ADN normal. La inestabilidad se observa mejor en
repeticiones de
mononucleótidos. Sensibilidad Reducida: Los marcadores actuales para el diagnóstico de MSI en
cáncer de colon
tienen sensibilidad reducida para detectar MSI en tumores no colónicos (endometrial, intestino
delgado,
gástrico) y en tumores de pacientes con mutaciones en MSH6.
Consideraciones en la Interpretación de dMMR e MSI:
Los tumores dMMR no siempre muestran inmunohistoquímica anormal o dan positivo para MSI. La
sensibilidad de IHQ y
MSI se reduce en carcinomas de sitios diferentes al colon, debido a mutaciones constitucionales
en
MSH6 o PMS2, o en adenomas. Debe interpretarse considerando el tipo de
tumor, el
historial personal y familiar del paciente, y otros resultados (ej. estado de
BRAF); se recomienda
un enfoque multidisciplinario.
Valor de la IHQ de MMR y MSI:
- Mejorado al analizar más de un tumor de la misma familia y/o individuo, especialmente si los tumores son raros (cánceres rectales, adenomas colorrectales, cánceres de intestino delgado, tumores cutáneos sebáceos).
- Una anomalía consistente en la IHQ de una proteína MMR es una excelente evidencia de patogenicidad de una mutación en ese gen.
- La MSI en cánceres rectales es rara y fuertemente asociada al SL, con un excelente valor predictivo positivo y negativo.
- La MSI en adenomas fuera del SL también es rara, proporcionando buenos valores predictivos.
- Algunos CRC debidos a poliposis asociada a
MUTYHo PPAP pueden mostrar MSI y IHQ anormal debido a mutaciones somáticas, aparentando ser SL (casos de"síndrome de Lynch-símil").
Distinción entre SL y Origen Esporádico:
- Aproximadamente el 15% de los cánceres de colon esporádicos tienen MSI, generalmente por
silenciamiento
epigenético de
MLH1por hipermetilación del promotor en ambos alelos. - Los CRC no seleccionados con MSI tienen un bajo valor predictivo positivo para SL, aunque la ausencia de MSI tiene un buen valor predictivo negativo.
- Se requieren pruebas adicionales (mutación
BRAFy metilación deMLH1) para cánceres microsatélite-inestables con pérdida deMLH1en inmunohistoquímica. - Las mutaciones en
BRAF(p.V600E) se encuentran en al menos el 85% de los CRC esporádicos con MSI, pero no en los debidos a SL; son altamente predictivas de origen esporádico. La ausencia de mutacionesBRAF p.V600Eno diagnostica definitivamente SL, pero indica mayor probabilidad. - La detección de hipermetilación del promotor de
MLH1en un tumor sugiere un origen esporádico, pero no de forma inequívoca. Es buena práctica incluir el ADN normal del paciente para esta prueba.
Diagnóstico Definitivo de SL:
- Después de las pruebas tumorales, se realiza la secuenciación constitucional del gen MMR para identificar la mutación patogénica.
- Pueden ser útiles muestras de varios individuos de la familia para estudios de segregación y determinación de patogenicidad.
- Si no se encuentra una mutación puntual, deben considerarse mutaciones a gran escala (deleciones).
- En casos de inicio muy temprano (<35 años) y sin antecedentes familiares, el SL es
probable, aunque deben
considerarse mutaciones en genes del punto de control del huso mitótico (ej.
BUB1yBUB3). - Algunos tumores pueden adquirir dos mutaciones en genes MMR debido a síndromes subyacentes no relacionados con SL (incluyendo PPAP), apareciendo como SL en las pruebas tumorales.
- Tumores raros característicos (cáncer de intestino delgado, carcinoma de células transicionales de uréter, adenoma/carcinoma sebáceo de piel) tienen un alto valor predictivo positivo para SL.
- Los cánceres de colon sincrónicos o metacrónicos, o el desarrollo de dos tumores relacionados con SL (ej. CRC y cáncer de endometrio), también son clínicamente significativos.
Impacto de la Deficiencia de Reparación de Errores en la Inmunidad:
Causa mutaciones frecuentes de inserción y deleción, generando péptidos frameshift antigénicos
que estimulan el
sistema inmunitario. Los inhibidores de puntos de control inmunitarios (ej. bloqueo de
PD1/PDL1)
son efectivos en tumores dMMR al inhibir la supresión inmunitaria local.
Futuro del Diagnóstico:
La secuenciación tumoral masiva y el análisis de espectros de mutaciones probablemente
reemplazarán las pruebas
de MSI, guiando el tratamiento y diagnosticando más causas de predisposición hereditaria
(incluyendo
"síndrome de Lynch-símil" y
síndrome de alelos de neoplasia heredada multilocus). La
inmunohistoquímica de MMR seguirá siendo crucial para proporcionar datos fenotípicos.
Criterios Diagnósticos Esenciales y Deseables
Se sugiere un algoritmo para las pruebas tumorales de CRC para SL.
Estadificación
No clínicamente relevante.
Pronóstico y Predicción
No clínicamente relevante.
Interpretación
Criterios generales
- Debe haber un mínimo de 100 células tumorales viables para la evaluación.
- Debe evitarse la puntuación en áreas necróticas.
- Cualquier grado de intensidad de tinción (1+ a 3+) cuenta para la puntuación.
- La puntuación es dependiente del sitio tumoral.
- La tinción puede estar presente tanto en el componente de células inmunes (IC) como en el de células tumorales (TC).
Células tumorales (TC)
- Solo debe evaluarse la tinción de membrana.
- Se incluye la tinción de membrana parcial o completa.
- No se incluye la tinción citoplasmática.
Células inmunes (IC)
- La tinción de membrana y citoplasmática suele ser indistinguible debido a la alta razón núcleo:citoplasma.
- Por lo tanto, se incluye tanto la tinción de membrana como la citoplasmática de las células inflamatorias.
- Solo se cuenta la tinción en linfocitos y macrófagos.
- No se cuenta la tinción en neutrófilos, eosinófilos ni células plasmáticas.
Carcinoma de pulmón de células no pequeñas (NSCLC)
Se evalúa mediante la puntuación de proporción tumoral (TPS).
TPS es el porcentaje de células tumorales viables que muestran tinción de membrana
parcial o completa (≥ 1+) en relación con todas las células tumorales viables presentes en la
muestra.
La muestra se considera que presenta expresión de PD-L1 si TPS ≥ 1 % y se
considera
alta expresión de PD-L1 si
TPS ≥ 50 %.
TPS = (número de células tumorales PDL1 positivas × 100) / (número total de células tumorales viables PDL1 positivas + PDL1 negativas)
Se divide en 3 niveles:
- TPS < 1 %: sin expresión de PD-L1.
- TPS 1 - 49 %: expresión de PD-L1.
- TPS ≥ 50 %: alta expresión de PD-L1.
Carcinoma cervical
La expresión se determina utilizando la puntuación positiva combinada (CPS).
CPS es el número de células con tinción de PD-L1 (células tumorales, linfocitos,
macrófagos) dividido por el número total de células tumorales viables, multiplicado por 100.
La muestra se considera que presenta expresión de PD-L1 si CPS ≥ 1.
Las células incluidas en el numerador son todas las células tumorales invasoras viables y las células inmunes mononucleares directamente asociadas a la respuesta al tumor (linfocitos y macrófagos dentro de los nidos tumorales y el estroma de sostén adyacente); las células inmunes mononucleares adyacentes se definen como aquellas que se encuentran en el mismo campo de 20× que el tumor.
CPS = (número de células con tinción de PD-L1 [células tumorales, linfocitos, macrófagos] × 100) / (número total de células tumorales viables)
El valor de CPS puede ser > 100, pero el valor máximo reportado es 100.
Se divide en 2 grupos:
- CPS < 1: sin expresión de PD-L1.
- CPS ≥ 1: expresión de PD-L1.
Adenocarcinoma gástrico o de la unión gastroesofágica (GEJ)
La expresión se determina utilizando CPS.
CPS es el número de células con tinción de PD-L1 (células tumorales, linfocitos,
macrófagos) dividido por el número total de células tumorales viables, multiplicado por 100.
La muestra se considera que presenta expresión de PD-L1 si CPS ≥ 1.
Las células incluidas en el numerador son todas las células tumorales invasoras viables y las células inmunes mononucleares directamente asociadas a la respuesta al tumor (linfocitos y macrófagos dentro de los nidos tumorales y el estroma de sostén adyacente); las células inmunes mononucleares adyacentes se definen como aquellas que se encuentran en el mismo campo de 20× que el tumor.
CPS = (número de células con tinción de PD-L1 [células tumorales, linfocitos, macrófagos] × 100) / (número total de células tumorales viables)
El valor de CPS puede ser > 100, pero el valor máximo reportado es 100.
Se divide en 2 grupos:
- CPS < 1: sin expresión de PD-L1.
- CPS ≥ 1: expresión de PD-L1.
Carcinoma escamoso de cabeza y cuello (HNSCC)
La expresión se determina utilizando CPS.
CPS es el número de células con tinción de PD-L1 (células tumorales, linfocitos,
macrófagos) dividido por el número total de células tumorales viables, multiplicado por 100.
La expresión de PD-L1 (CPS ≥ 1) se utiliza para informar la elegibilidad del
paciente para terapia de primera línea con pembrolizumab.
Las células incluidas en el numerador son todas las células tumorales invasoras viables y las células inmunes mononucleares directamente asociadas a la respuesta al tumor (linfocitos y macrófagos dentro de los nidos tumorales y el estroma de sostén adyacente); las células inmunes mononucleares adyacentes se definen como aquellas que se encuentran en el mismo campo de 20× que el tumor.
Se divide en 3 grupos:
- CPS < 1: sin expresión de PD-L1.
- CPS ≥ 1: expresión de PD-L1.
- CPS ≥ 20: expresión de PD-L1 elevada.
CPS ≥ 20 puede ser de interés para los
médicos tratantes, pero no determina la elegibilidad para terapia de primera línea.
Carcinoma escamoso de esófago (ESCC)
La expresión se determina utilizando CPS.
CPS es el número de células con tinción de PD-L1 (células tumorales, linfocitos,
macrófagos) dividido por el número total de células tumorales viables, multiplicado por 100.
La muestra se considera que presenta expresión de PD-L1 si CPS ≥ 10.
Las células incluidas en el numerador son todas las células tumorales invasoras viables y las células inmunes mononucleares directamente asociadas a la respuesta al tumor (linfocitos y macrófagos dentro de los nidos tumorales y el estroma de sostén adyacente); las células inmunes mononucleares adyacentes se definen como aquellas que se encuentran en el mismo campo de 20× que el tumor.
Se divide en 2 grupos:
- CPS < 10: sin expresión de PD-L1.
- CPS ≥ 10: expresión de PD-L1.
Cáncer de mama triple negativo (TNBC)
La expresión se determina utilizando CPS.
CPS es el número de células con tinción de PD-L1 (células tumorales, linfocitos,
macrófagos) dividido por el número total de células tumorales viables, multiplicado por 100.
La muestra se considera que presenta expresión de PD-L1 si CPS ≥ 10.
Se divide en 2 grupos:
- CPS < 10.
- CPS ≥ 10.
Carcinoma urotelial
Referencias
Agilent: PD-L1 IHC 22C3 pharmDx Overview [consultado el 2 de febrero de 2023],
Agilent: PD-L1 IHC 22C3 pharmDx Interpretation Manual - NSCLC [consultado el 2 de
marzo de 2023],
Dako:
PD-L1 IHC 22C3 pharmDx Interpretation Manual - Esophageal Squamous Cell Carcinoma (ESCC)
[consultado el 16 de enero de 2023].
Tipos de células gigantes en patología
1. Células gigantes derivadas de macrófagos
A. Células gigantes tipo Langhans
Las células gigantes tipo Langhans presentan los núcleos dispuestos en forma de herradura en un polo de la célula y suelen contener más de 15 núcleos.
Se observan en:
- Granuloma tuberculoso
- Lepra
- Sífilis
- Infecciones micóticas profundas
- Sarcoidosis
- Leishmaniasis
- Enfermedad de Crohn
B. Células gigantes de cuerpo extraño
Las células gigantes de cuerpo extraño son más grandes que las tipo Langhans y muestran los núcleos dispersos al azar en el citoplasma.
Se observan en:
- Granulomas por cuerpo extraño formados en respuesta a materiales exógenos o endógenos.
C. Células gigantes de Touton
Las células gigantes de Touton tienen un anillo central de núcleos, mientras que el citoplasma periférico es claro por la acumulación de lípidos.
Se observan en:
- Necrosis grasa
- Xanthoma
- Xanthogranulomas
- Dermatofibroma
2. Células gigantes derivadas de células epidérmicas
A. Células de Tzanck
Las células de Tzanck son células gigantes multinucleadas con moldeamiento nuclear, ya que los núcleos están apiñados entre sí.
Se observan en:
- Herpes simplex
- Varicela y herpes zóster
- Cytomegalovirus
B. Células epidérmicas gigantes multinucleadas
Las células epidérmicas gigantes multinucleadas (con más de tres núcleos agrupados) se consideran un fenómeno episódico y excepcional en enfermedades inflamatorias de la piel.
Se observan en:
- Eccema crónico o prurigo
- Lichen amyloidosis
- Dermatitis herpetiformis
- Erythema multiforme
- Psoriasis pustulosa
- Lichen planus
- Lupus erythematosus
3. Células gigantes derivadas de melanocitos
A. Células gigantes tipo “starburst”
Las células gigantes tipo starburst son melanocitos multinucleados con aspecto estrellado por sus prolongaciones dendríticas prominentes. Son un indicador útil para el diagnóstico de lentigo maligna.
B. Células en balón
Las células en balón pueden ser multinucleadas y son más grandes que las células névicas ordinarias. Se observan en balloon cell nevus y en melanoma de células en balón.
4. Células gigantes multinucleadas tipo “floret”
Las células gigantes multinucleadas tipo floret presentan citoplasma escaso y núcleos dispuestos en la periferia, dentro del estroma interpuesto.
Se observan en:
- Ginecomastia
- Neurofibroma en pacientes con NF-1
- Giant cell angiofibroma
- Lipoma pleomórfico
5. Células gigantes asociadas a tumores
- Células gigantes de Reed-Sternberg en linfoma de Hodgkin y en otros neoplasmas hematolinfoides.
- Células gigantes tipo osteoclasto en tumores de células gigantes del hueso y en otras lesiones óseas.
- Células gigantes en ganglio linfático asociadas a linfoma anaplásico de células grandes.
Adaptado de www.pathologymcq.com.
Adenocarcinoma Ampular
Definición
El adenocarcinoma ampular es un tumor epitelial maligno del intestino delgado, con su epicentro en la ampolla, que muestra diferenciación glandular o mucinosa.
Codificación ICD-O
- 8140/3 Adenocarcinoma NOS
Codificación ICD-11
- 2C16.0 Adenocarcinoma de la ampolla de Vater
Terminología Relacionada
Ninguna
Subtipo(s)
- Mucinous adenocarcinoma (8480/3)
- Signet-ring cell carcinoma (8490/3)
- Poorly cohesive carcinoma (8490/3)
- Medullary carcinoma NOS (8510/3)
- Adenocarcinoma, intestinal type (8144/3)
- Pancreatobiliary-type carcinoma (8163/3)
- Tubular adenocarcinoma (8211/3)
- Adenosquamous carcinoma (8560/3)
- Carcinoma, undifferentiated, NOS (8020/3)
Localización
La ampolla es la localización más común para el adenocarcinoma del intestino delgado { 9261617 }, atribuido a los efluentes biliares y/o pancreáticos. Para establecer si un tumor es de origen ampular (y para excluir tumores metastásicos de las regiones inmediatamente vecinas del conducto biliar y el páncreas), es crucial una disección adecuada de los especímenes de pancreatoduodenectomía (para documentar que el epicentro del cáncer está en la ampolla). El enfoque de bivalvado { 24451278 }, con seccionamiento del conducto biliar común y el conducto pancreático principal a lo largo del mismo plano en el que entran a la ampolla, permite la demostración de la localización específica del tumor, así como la subclasificación adicional de los tumores ampulares según el sitio específico { 23026934 ; 23026934 }.
Los adenocarcinomas ampulares pueden surgir de (y crecer preferentemente en) los diversos compartimentos de este pequeño órgano.
- Carcinomas (peri)ampulares-duodenales: Son aquellos que surgen de la superficie duodenal de la ampolla (a menudo de un adenoma) y característicamente forman masas ulcerantes o vegetantes que son fácilmente visibles desde la perspectiva duodenal. En los tumores (peri)ampulares-duodenales, el orificio de la ampolla típicamente está englobado excéntricamente en el polo superior de la lesión.
-
Carcinomas intra-ampulares: Desde la vista duodenal, aparecen solo como úlceras sutiles, elevaciones cubiertas de mucosa o material granular que protruye desde dentro de la ampolla. Ocurren en dos subconjuntos distintos según la localización:
- Surgidos de la neoplasia papilar-tubular intra-ampular y caracterizados por una masa polipoide, nodular o granular localizada dentro de la cavidad ampular.
- Surgidos de los conductos ampulares, con un tumor escirroso, constrictivo y circunferencial localizado en las paredes de los conductos ampulares (los extremos distales del conducto biliar común y la ampolla).
- Carcinoma ampular NOS: Es el diagnóstico reservado para tumores claramente de origen ampular pero que no pueden ser clasificados en ninguna de las tres categorías anteriores, a pesar del manejo adecuado de los especímenes y la correlación macro-microscópica.
Características Clínicas
A diferencia de los adenocarcinomas en otros segmentos del intestino delgado, los adenocarcinomas ampulares a menudo se detectan en una etapa relativamente temprana debido a los síntomas y signos causados por la obstrucción de los conductos biliares, lo que conduce a colestasis y hallazgos relacionados. Para el subconjunto de casos (peri)ampulares-duodenales, que a menudo forman úlceras, la presentación inicial suele ser sangrado gastrointestinal con sangre oculta. Ocasionalmente, también puede ocurrir lesión/insuficiencia pancreática debido a la oclusión del flujo pancreático, pero los hallazgos suelen estar más relacionados con la obstrucción del conducto biliar { 23026934 ; 20871215 ; 21084962 ; 11982470 }. Cerca de la mitad de los casos presentan metástasis en los ganglios linfáticos en el momento de la resección { 25783680 }.
Epidemiología
El adenocarcinoma ampular representa el 6–9% de todas las malignidades periampulares. Aproximadamente el 15% de las pancreatoduodenectomías se realizan por cánceres ampulares { 23026934 ; 25783680 ; 20871215 ; 29518820 }. Los hombres se ven afectados con una frecuencia ligeramente mayor que las mujeres, y la edad media de los pacientes está a mediados de los 60 años { 23026934 ; 25783680 ; 21084962 }.
Etiología
Para la gran mayoría de los adenocarcinomas ampulares, no es evidente una etiología específica { 23026934 ; 21084962 ; 23439753 }. Algunas de las enfermedades inflamatorias (p. ej., Crohn, celíaca) y procedimientos quirúrgicos previos, que han sido documentados como factores predisponentes importantes para los adenocarcinomas de otras partes del intestino delgado, representan solo un pequeño porcentaje de los casos en la ampolla.
Las condiciones hereditarias como la poliposis adenomatosa familiar, el síndrome de Lynch y el síndrome de Peutz-Jeghers también son asociaciones relevantes.
Patogénesis
Se cree que la predilección de los adenocarcinomas del intestino delgado por ocurrir en la región ampular se atribuye a la exposición a la bilis y los productos pancreáticos. La ampolla también es uno de los sitios comunes para los adenocarcinomas relacionados con el síndrome de poliposis adenomatosa familiar, aunque tales casos representan un porcentaje muy pequeño de los cánceres ampulares. Además, se han descrito mutaciones específicas y una firma de expresión génica con potencial significado clínico en este cáncer (ver Pronóstico y predicción, más abajo).
Apariencia Macroscópica
La apariencia macroscópica de estos carcinomas depende de la porción de la ampolla de la que derivan.
- Carcinomas que surgen en el duodeno (peri)ampular: Típicamente se presentan como grandes masas ulcerovegetantes que son evidentes al abrir el duodeno e inspeccionar esta región. Los componentes nodulares polipoides corresponden a adenomas, y las áreas ulceradas (que pueden estar ocultas en los pliegues debajo de las áreas polipoides y, por lo tanto, no ser detectadas por biopsias) a menudo revelan carcinomas invasivos. En los cánceres (peri)ampulares-duodenales, el orificio ampular puede estar oscurecido y ser difícil de localizar, pero las sondas insertadas en el conducto biliar común proximal a menudo salen de forma excéntrica y superior en esta masa.
- Carcinomas que surgen de neoplasias papilares-tubulares intra-ampulares: Por definición, tienen material granular nodular polipoide que llena la cavidad ampular. Desde la perspectiva duodenal, estas neoplasias pueden presentarse como protuberancias cubiertas de mucosa, a menudo con un orificio ampular dilatado ubicado centralmente, desde el cual puede protruir material nodular granular hacia el lumen duodenal.
- Carcinomas de origen en los conductos ampulares: También son «intra-ampulares» y, desde la perspectiva duodenal, a menudo aparecen como elevaciones en forma de botón cubiertas de mucosa en la ampolla, algunas con ulceración sutil. El bivalvado de la ampolla típicamente revela un engrosamiento esclerosante mal definido de los conductos intra-ampulares que conduce a lesiones constrictivas circunferenciales.
Histopatología
La gran mayoría de los adenocarcinomas ampulares, independientemente de la clasificación específica del sitio, son adenocarcinomas formadores de glándulas (tubulares) { 27739441 ; 20334897 ; 22187121 }.
Históricamente, se esperaba que los adenocarcinomas ampulares se clasificaran en una de dos categorías histológicas amplias: pancreatobiliar o intestinal (tipo colónico). Sin embargo, alrededor del 40% de los casos revelan fenotipos mixtos o híbridos, lo que también se refleja en los inmunoperfiles { 27739441 ; 27586202 }. Esto también deja un número sustancial de casos con inmunoperfiles heterogéneos { 24832159 ; 23439753 ; 28505002 ; 21084962 }. Esto no es sorprendente considerando que la ampolla es una región de transición con compartimentos celulares distintivos, así como su exposición a diferentes factores etiopatogénicos, junto con el hecho de que el epitelio del intestino delgado no es verdaderamente colónico, y los conductos ampulares no son exactamente idénticos a los conductos pancreatobiliares.
No obstante, los paneles de tinciones inmunohistoquímicas que incluyen EMA (MUC1), MUC2, CDX2 y MUC5AC pueden ayudar en la clasificación de los adenocarcinomas con patrón tubular como tipo intestinal o pancreatobiliar en una proporción sustancial de casos { 24832159 ; 23439753 ; 28505002 }.
Adenocarcinoma de tipo intestinal
Estos adenocarcinomas formadores de glándulas (túbulos) son morfológicamente similares a los adenocarcinomas de colon { 27586202 }. Las glándulas están revestidas por células columnares altas con núcleos alargados y pseudoestratificados. Pueden estar presentes células caliciformes, de Paneth y neuroendocrinas dispersas { 27586202 ; 7076140 ; 1929795 }. Una proporción sustancial está asociada con adenoma de tipo intestinal { 7076140 ; 6691630 ; 1778589 }. En la región ampular especialmente, algunos de estos tumores exhiben otros patrones e inmunofenotipos diferentes a los de los verdaderos adenocarcinomas de tipo intestinal colónico (ver más abajo) { 27586202 }.
Adenocarcinoma de tipo pancreatobiliar o gástrico
Similar a los adenocarcinomas ductales pancreáticos, de vesícula biliar y del tracto biliar extrahepático, este tipo está compuesto por unidades glandulares relativamente pequeñas ampliamente separadas por estroma desmoplásico. El epitelio suele ser una sola capa de células cuboidales a columnares bajas, sin una pseudoestratificación nuclear sustancial, pero con más pleomorfismo que en el tipo intestinal { 27586202 ; 23026934 ; 18087285 }. Aunque los tumores con mucina de tipo gástrico (histológica e inmunofenotípicamente) han sido designados como tipo gástrico { 28505002 }, los dos grupos parecen estar estrechamente relacionados y a menudo se consideran un solo grupo { 28505002 ; 21084962 }.
Adenocarcinoma tubular con características mixtas
Muchos carcinomas en la ampolla tienen características ambiguas que son difíciles de clasificar definitivamente como pancreatobiliares o intestinales. Estos casos ambiguos/híbridos representan más de un tercio de todos los carcinomas ampulares { 24832159 ; 27739441 ; 27586202 ; 28505002 } y deben clasificarse como adenocarcinomas tubulares con características mixtas, señalando el patrón predominante. Los estudios inmunohistoquímicos con EMA (MUC1), MUC2, CDX2, CK20 y MUC5AC pueden ser útiles para determinar el linaje predominante { 27739441 ; 27586202 ; 28505002 ; 24832159 }.
Patrones no tubulares
Además de los tipos de adenocarcinoma convencionales, la mayoría de los cuales tienen un patrón tubular, también pueden ocurrir en esta región carcinomas con patrones no glandulares, pero a menudo se asocian con adenocarcinomas formadores de túbulos ordinarios. En algunos casos, los patrones no tubulares pueden predominar y los tumores se clasifican en consecuencia. Estos se discuten a continuación.
Mucinous adenocarcinoma
El adenocarcinoma mucinoso (con > 50% de depósito de mucina estromal) es proporcionalmente más común en el duodeno. En la ampolla, se observa cierto grado de depósito de mucina extracelular en el 15% de los casos, y dos tercios califican como adenocarcinoma mucinoso { 19697352 }. Las tiras y glándulas epiteliales malignas que flotan en lagos de mucina suelen ser columnares con fenotipo intestinal (también con expresión difusa de CDX2 y MUC2), lo que acerca estos casos a los adenocarcinomas de tipo intestinal. Se pueden observar células en anillo de sello dispersas flotando dentro de la mucina, pero no infiltrando el estroma como células individuales o cordones, una distinción importante con el verdadero carcinoma de células poco cohesivas. El adenocarcinoma mucinoso en la ampolla surge más comúnmente de adenomas del duodeno ampular, pero también puede verse en asociación con tumores intra-ampulares.
Carcinoma de células poco cohesivas
Estos tumores, que son relativamente poco comunes en el intestino delgado, a menudo carecen de un componente adenomatoso { 19697352 ; 26057354 ; 25202392 }, suelen estar avanzados en el momento del diagnóstico y se comportan de manera más agresiva.
Carcinoma medular
Los carcinomas medulares, similares a sus contrapartes colónicas, pueden ocurrir en el intestino delgado, especialmente en el duodeno y la ampolla { 27739441 }. Constituyen el 3% de los cánceres ampulares y se caracterizan por masas grandes y nodulares con bordes de empuje y crecimiento sincitial, con inflamación asociada. Al igual que en el colon, se comportan de manera menos agresiva a pesar de su gran tamaño.
Carcinoma adenoescamoso
El carcinoma adenoescamoso es extremadamente raro. Representa el 1% de todos los carcinomas ampulares { 16929159 ; 26420726 ; 23721111 }. Un muestreo adecuado a menudo ilustra el componente glandular. El componente escamoso exhibe citoqueratinas de tipo escamoso, junto con p63 y p40. Un carcinoma de células escamosas puro debería plantear la posibilidad de metástasis.
Carcinoma neuroendocrino de alto grado (NEC) y carcinomas mixtos
Los NEC pobremente diferenciados (tanto de células pequeñas como de células grandes) pueden surgir en el intestino delgado, particularmente en la ampolla { 15832081 ; 11706080 ; 15747032 ; 15223956 }. Se justifica la confirmación inmunohistoquímica de la diferenciación neuroendocrina con sinaptofisina y cromogranina A, ya que otras neoplasias malignas poco diferenciadas pueden tener características similares a las neuroendocrinas (ver sección Neoplasias neuroendocrinas del intestino delgado y ampulares).
Carcinoma indiferenciado
- Con células gigantes tipo osteoclasto: El carcinoma indiferenciado con células gigantes tipo osteoclasto, como se describe en el páncreas, puede ocurrir ocasionalmente en el intestino delgado, más a menudo en la ampolla o el duodeno { 27508975 ; 27281360 ; 7611227 ; 20339995 }. Debe excluirse la afectación metastásica o directa por un primario de páncreas o conducto biliar { 27508975 }. Las células gigantes en estos tumores son osteoclastos convencionales (histiocíticos y no neoplásicos) y marcan positivamente para el marcador histiocítico CD68. En contraste, las células mononucleares neoplásicas, que a menudo tienen una apariencia sarcomatoide, pueden ser positivas para queratina y a menudo muestran un patrón de tinción de p53 mutante { 9777987 }.
- Con fenotipo rabdoide: Se han descrito ejemplos muy raros de carcinomas indiferenciados con un fenotipo rabdoide distintivo en la ampolla y el intestino delgado { 26551623 }. Los tumores son típicamente grandes y muestran células rabdoides características con abundantes cuerpos rabdoides eosinofílicos intracitoplasmáticos. Las células tumorales a menudo son discohesivas y están incrustadas en una matriz mixoide. También se puede observar diferenciación pleomórfica de células gigantes o fusiformes, así como áreas de diferenciación glandular. La pérdida de la inmunotinción nuclear para SMARCB1 (INI1), una subunidad central del complejo de remodelación de la cromatina SWI/SNF, es característica. Estos tumores parecen tener pérdida/coactivación de múltiples miembros de la familia de proteínas SWI/SNF, incluyendo SMARCA4, SMARCA2 y/o SMARCB1, lo que indica una asociación entre la deficiencia de SWI/SNF y el fenotipo rabdoide. Algunos tienen anomalías en ARID1A.
Citología
Para los casos con un gran componente adenomatoso y áreas infiltrativas más profundas en los tejidos, la PAAF (FNA) puede ser informativa, pero rara vez se utiliza, ya que la mayoría de los tumores ampulares son fácilmente accesibles por endoscopia duodenal en lugar de requerir PAAF { 11376018 }.
Patología Molecular Diagnóstica
Se han identificado mutaciones en KRAS que muestran un valor pronóstico potencial, pero tienen poco valor diagnóstico y ningún valor predictivo { 27517148 ; 26997442 }. Más recientemente, se ha postulado una firma de 92 genes que tiene una concordancia diagnóstica significativa con el estándar de oro de clasificación, y tiene un valor pronóstico objetivo, particularmente en casos poco diferenciados { 27549176 }.
La inestabilidad de microsatélites parece estar involucrada en aproximadamente el 15% de los carcinomas ampulares, y el análisis inmunohistoquímico para las proteínas de reparación de desajustes (mismatch repair) puede ser útil.
Criterios Diagnósticos Esenciales y Deseables
- Esencial: Adenocarcinoma de origen ampular con diferenciación principalmente glandular y raramente escamosa, neuroendocrina o sarcomatoide.
Estadificación
El adenocarcinoma ampular se estadifica patológicamente según la octava edición (2017) de la clasificación TNM de la Union for International Cancer Control (UICC) [[Brierly JD, Gospodarowicz MK, Wittekind C, editors. TNM classification of malignant tumours. 8th ed. Oxford (UK): Wiley Blackwell; 2017.]]. Sin embargo, debido a la complejidad anatómica de los cánceres ampulares, es crucial un examen macroscópico y una disección adecuados de los especímenes de resección para lograr una estadificación correcta { 24451278 }.
Es crítico un examen cuidadoso de cualquier extensión a los tejidos blandos periduodenales adyacentes a la ampolla (donde no está amortiguada por el páncreas), ya que los carcinomas infiltran fácilmente esta región sin penetrar primero a través del páncreas { 23062420 }.
Pronóstico y Predicción
Los carcinomas ampulares resecados tienen una tasa de supervivencia global a 5 años del 45% { 23026934 ; 19697352 ; 18369675 ; 20871215 }. Los factores pronósticos independientes incluyen la edad del paciente, metástasis en ganglios linfáticos, invasión perineural y vascular, estado de los márgenes, tumour budding y expresión de MUC5AC { 28505002 }; el tamaño del carcinoma invasivo, el tipo histológico y el grado parecen jugar un papel menor { 23026934 ; 25783680 ; 20871215 }.
Los carcinomas de origen ductal-ampular suelen ser agresivos a pesar de su pequeño tamaño; presumiblemente son de tipo pancreatobiliar y de estadio T avanzado debido a su fácil acceso al tejido blando { 23026934 ; 21084962 }.
Es de destacar que el fenotipo pancreatobiliar en el carcinoma ampular tiene un pronóstico sustancialmente mejor que su contraparte pancreática (adenocarcinoma ductal pancreático), y no es un factor independiente por sí mismo cuando se compara con el fenotipo intestinal { 27586202 }.
Consulte la subsección de Patología Molecular Diagnóstica para una discusión sobre el uso de pruebas moleculares con valor terapéutico predictivo.
Marcadores de Inmunohistoquímica
En la siguiente tabla se organizan los enlaces por categoría y subcategoría. Todos los enlaces se abren en una nueva pestaña.
| Categoría | Subcategoría | Marcador / recurso |
|---|---|---|
| Hodgkin lymphoma | ALK1 | |
| Hodgkin lymphoma | CD3 | |
| Hodgkin lymphoma | CD15 | |
| Hodgkin lymphoma | CD20 | |
| Hodgkin lymphoma | CD30 | |
| Hodgkin lymphoma | CD45 (LCA) | |
| Hodgkin lymphoma | EBER1 / 2 / LMP | |
| Hodgkin lymphoma | PAX5 | |
| Leukemia | CD3 | |
| Leukemia | CD34 | |
| Leukemia | CD68 | |
| Leukemia | c-kit / CD117 | |
| Leukemia | E-cadherin | |
| Leukemia | MPO | |
| Leukemia | NPM1 | |
| Leukemia | PAX5 | |
| Leukemia | TdT | |
| Non-Hodgkin lymphoma & myeloma | BCL6 | |
| Non-Hodgkin lymphoma & myeloma | CD3 | |
| Non-Hodgkin lymphoma & myeloma | CD5 | |
| Non-Hodgkin lymphoma & myeloma | CD10 | |
| Non-Hodgkin lymphoma & myeloma | CD15 | |
| Non-Hodgkin lymphoma & myeloma | CD20 | |
| Non-Hodgkin lymphoma & myeloma | CD23 | |
| Non-Hodgkin lymphoma & myeloma | CD30 | |
| Non-Hodgkin lymphoma & myeloma | CXCR3 | |
| Non-Hodgkin lymphoma & myeloma | kappa & lambda light chains | |
| Non-Hodgkin lymphoma & myeloma | Ki67 | |
| Nonneoplastic lymph node & bone marrow | AFB | |
| Nonneoplastic lymph node & bone marrow | CD1a | |
| Nonneoplastic lymph node & bone marrow | CD34 | |
| Nonneoplastic lymph node & bone marrow | CD41 | |
| Nonneoplastic lymph node & bone marrow | CD45 (LCA) | |
| Nonneoplastic lymph node & bone marrow | CD61 | |
| Nonneoplastic lymph node & bone marrow | CD68 | |
| Nonneoplastic lymph node & bone marrow | c-kit / CD117 | |
| Nonneoplastic lymph node & bone marrow | CMV | |
| Nonneoplastic lymph node & bone marrow | E-cadherin | |
| Nonneoplastic lymph node & bone marrow | EBER1 / 2 / LMP | |
| Nonneoplastic lymph node & bone marrow | EMA | |
| Nonneoplastic lymph node & bone marrow | Factor VIII | |
| Nonneoplastic lymph node & bone marrow | GMS | |
| Nonneoplastic lymph node & bone marrow | HHV8 | |
| Nonneoplastic lymph node & bone marrow | IgG4 | |
| Nonneoplastic lymph node & bone marrow | iron | |
| Nonneoplastic lymph node & bone marrow | myeloperoxidase | |
| Nonneoplastic lymph node & bone marrow | PAS | |
| Nonneoplastic lymph node & bone marrow | reticulin | |
| Nonneoplastic lymph node & bone marrow | S100 | |
| Nonneoplastic lymph node & bone marrow | trichrome | |
| Nonneoplastic lymph node & bone marrow | Warthin-Starry | |
| Lung & pleura | ALK | |
| Lung & pleura | calretinin | |
| Lung & pleura | chromogranin | |
| Lung & pleura | CK5/6 and CK5 | |
| Lung & pleura | CK7 | |
| Lung & pleura | D2-40 | |
| Lung & pleura | EGFR mutation specific antibodies | |
| Lung & pleura | Napsin A | |
| Lung & pleura | OTP (orthopedia) (pending) | |
| Lung & pleura | PDL1 22C3 | |
| Lung & pleura | PDL1 SP142 | |
| Lung & pleura | synaptophysin | |
| Lung & pleura | TTF1 | |
| Lung & pleura | WT1 | |
| Medical renal | adenovirus | |
| Medical renal | C4d (pending) | |
| Medical renal | calcium / von Kossa | |
| Medical renal | CD68 | |
| Medical renal | CMV | |
| Medical renal | collagen / type IV | |
| Medical renal | Congo red | |
| Medical renal | COX2 | |
| Medical renal | DNAJB9 (pending) | |
| Medical renal | EBER1 / 2 / LMP | |
| Medical renal | FN1 (pending) | |
| Medical renal | HSV (pending) | |
| Medical renal | Jones silver | |
| Medical renal | Masson trichrome | |
| Medical renal | myogenin / myoglobin | |
| Medical renal | NCCT (pending) | |
| Medical renal | PAX2 | |
| Medical renal | Periodic acid Schiff (PAS) | |
| Medical renal | prealbumin (pending) | |
| Medical renal | SV40 (pending) | |
| Neuropath | CNS nontumor | alpha-synuclein |
| Neuropath | CNS nontumor | AT8 (pending) |
| Neuropath | CNS nontumor | beta amyloid |
| Neuropath | CNS nontumor | Luxol fast blue |
| Neuropath | CNS nontumor | p62 (pending) |
| Neuropath | CNS nontumor | TDP-43 (pending) |
| Neuropath | CNS tumor | ACTH (pending) |
| Neuropath | CNS tumor | ATRX |
| Neuropath | CNS tumor | BRAF V600E |
| Neuropath | CNS tumor | H3K27M (pending) |
| Neuropath | CNS tumor | H3K36M (pending) |
| Neuropath | CNS tumor | GFAP |
| Neuropath | CNS tumor | IDH1 (R132H) |
| Neuropath | CNS tumor | NeuN |
| Neuropath | CNS tumor | NFP / neurofilament |
| Neuropath | CNS tumor | Olig2 |
| Neuropath | CNS tumor | Pit1 |
| Neuropath | CNS tumor | SSTR2A |
| Neuropath | CNS tumor | synaptophysin |
| Neuropath | CNS tumor | Tpit |
| Neuropath | Eye | PASD |
| Neuropath | Muscle nontumor | acid phosphatase |
| Neuropath | Muscle nontumor | alkaline phosphatase |
| Neuropath | Muscle nontumor | COX |
| Neuropath | Muscle nontumor | NADH |
| Neuropath | Muscle nontumor | nonspecific esterase |
| Neuropath | Muscle nontumor | Oil Red O |
| Neuropath | Muscle nontumor | PAS |
| Neuropath | Muscle nontumor | SDH (pending) |
| Companion diagnostic tests | c-kit / CD117 | |
| Companion diagnostic tests | EGFR mutation specific antibodies | |
| Companion diagnostic tests | estrogen receptor (ER) | |
| Companion diagnostic tests | HER2 breast | |
| Companion diagnostic tests | HER2 colon | |
| Companion diagnostic tests | HER2 stomach GEJ | |
| Companion diagnostic tests | PDL1 22C3 | |
| Companion diagnostic tests | PDL1 SP142 | |
| Companion diagnostic tests | PDL1 SP263 (pending) | |
| Companion diagnostic tests | progesterone receptor (PR) | |
| Companion diagnostic tests | TROP2 (pending) | |
| Alphabetical listing | General | books |
| Alphabetical listing | General | IHC procedure |
| Alphabetical listing | General | common panels / unknown primary |
| Alphabetical listing | General | enzyme cytochemistry |
| Alphabetical listing | General | flow cytometry |
| Alphabetical listing | General | 2-succino-cysteine |
| Alphabetical listing | General | ABCC2 |
| Alphabetical listing | General | acid fast |
| Alphabetical listing | General | acid phosphatase |
| Alphabetical listing | General | ACTH (pending) |
| Alphabetical listing | General | actin-general / cardiac |
| Alphabetical listing | General | actin-alpha smooth muscle |
| Alphabetical listing | General | actin-muscle specific |
| Alphabetical listing | General | adenovirus |
| Alphabetical listing | General | adipophilin (pending) |
| Alphabetical listing | General | AE1/AE3 (cytokeratin) |
| Alphabetical listing | General | albumin |
| Alphabetical listing | General | Alcian blue |
| Alphabetical listing | General | ALK |
| Alphabetical listing | General | alkaline phosphatase |
| Alphabetical listing | General | alpha-1-antichymotrypsin |
| Alphabetical listing | General | alpha-1 antitrypsin |
| Alphabetical listing | General | alpha-fetoprotein (AFP) |
| Alphabetical listing | General | alpha-synuclein |
| Alphabetical listing | General | AMACR |
| Alphabetical listing | General | amyloid beta / APP |
| Alphabetical listing | General | androgen receptor (AR) |
| Alphabetical listing | General | arginase1 |
| Alphabetical listing | General | ARID1A |
| Alphabetical listing | General | AT8 (pending) |
| Alphabetical listing | General | ATDX (ATRX) |
| Alphabetical listing | General | ATM |
| Alphabetical listing | General | auramine-rhodamine |
| Alphabetical listing | General | B72.3 |
| Alphabetical listing | General | BAP1 |
| Alphabetical listing | General | BCL1 |
| Alphabetical listing | General | BCL10 (pending) |
| Alphabetical listing | General | BCL2 |
| Alphabetical listing | General | BCL6 |
| Alphabetical listing | General | BCOR |
| Alphabetical listing | General | BerEP4 |
| Alphabetical listing | General | beta catenin |
| Alphabetical listing | General | BOB1 |
| Alphabetical listing | General | brachyury |
| Alphabetical listing | General | BRAF |
| Alphabetical listing | General | BRAF-melanoma |
| Alphabetical listing | General | BSEP (pending) |
| Alphabetical listing | General | C4d (pending) |
| Alphabetical listing | General | CA 19-9 |
| Alphabetical listing | General | CA125 |
| Alphabetical listing | General | calcitonin |
| Alphabetical listing | General | calcium |
| Alphabetical listing | General | caldesmon |
| Alphabetical listing | General | calponin |
| Alphabetical listing | General | calretinin |
| Alphabetical listing | General | CAMTA1 |
| Alphabetical listing | General | carbonic anhydrase IX / CAIX |
| Alphabetical listing | General | cathepsin K |
| Alphabetical listing | CD markers | CD markers-general |
| Alphabetical listing | CD markers | 1-9 |
| Alphabetical listing | CD markers | 10-19 |
| Alphabetical listing | CD markers | 20-29 |
| Alphabetical listing | CD markers | 30-39 |
| Alphabetical listing | CD markers | 40-49 |
| Alphabetical listing | CD markers | 50-59 |
| Alphabetical listing | CD markers | 60-69 |
| Alphabetical listing | CD markers | 70-79 |
| Alphabetical listing | CD markers | 80-89 |
| Alphabetical listing | CD markers | 90-99 |
| Alphabetical listing | CD markers | 100-109 |
| Alphabetical listing | CD markers | 110-119 |
| Alphabetical listing | CD markers | 120-129 |
| Alphabetical listing | CD markers | 130-139 |
| Alphabetical listing | CD markers | 140-149 |
| Alphabetical listing | CD markers | 150-159 |
| Alphabetical listing | CD markers | 160-169 |
| Alphabetical listing | CD markers | 170-179 |
| Alphabetical listing | CD markers | 180-189 |
| Alphabetical listing | CD markers | 190-199 |
| Alphabetical listing | CD markers | 200-209 |
| Alphabetical listing | CD markers | 210-219 |
| Alphabetical listing | CD markers | 220-229 |
| Alphabetical listing | CD markers | 230-239 |
| Alphabetical listing | CD markers | 240-249 |
| Alphabetical listing | CD markers | 270-279 |
| Alphabetical listing | CD markers | 280-289 |
| Alphabetical listing | CD markers | 350-359 |
| Alphabetical listing | CD markers | 360-369 |
| Alphabetical listing | CD markers | 370-371 |
| Alphabetical listing | General | CDK4 |
| Alphabetical listing | General | CDX2 |
| Alphabetical listing | General | CEA |
| Alphabetical listing | General | chloroacetate esterase |
| Alphabetical listing | General | chromogranin |
| Alphabetical listing | General | c-kit / CD117 |
| Alphabetical listing | General | claudins |
| Alphabetical listing | General | CMV |
| Alphabetical listing | General | collagen / type IV |
| Alphabetical listing | General | Congo red |
| Alphabetical listing | General | copper / rhodamine |
| Alphabetical listing | General | COX |
| Alphabetical listing | General | COX2 |
| Alphabetical listing | General | CXCL13 (pending) |
| Alphabetical listing | General | CXCR3 |
| Alphabetical listing | General | Cyclin D1 |
| Alphabetical listing | Cytokeratins | general |
| Alphabetical listing | Cytokeratins | uncommon |
| Alphabetical listing | Cytokeratins | CK5/6 and CK5 |
| Alphabetical listing | Cytokeratins | CK7 |
| Alphabetical listing | Cytokeratins | CK8 |
| Alphabetical listing | Cytokeratins | CK8/18 (pending) |
| Alphabetical listing | Cytokeratins | CK10 |
| Alphabetical listing | Cytokeratins | CK14 |
| Alphabetical listing | Cytokeratins | CK17 |
| Alphabetical listing | Cytokeratins | CK18 |
| Alphabetical listing | Cytokeratins | CK19 |
| Alphabetical listing | Cytokeratins | CK20 |
| Alphabetical listing | Cytokeratins | 34betaE12 / HMWCK / high molecular weight |
| Alphabetical listing | Cytokeratins | 35betaH11 |
| Alphabetical listing | Cytokeratins | AE1/AE3 |
| Alphabetical listing | Cytokeratins | CAM5.2 |
| Alphabetical listing | Cytokeratins | MNF116 |
| Alphabetical listing | Cytokeratins | OSCAR |
| Alphabetical listing | General | D2-40 |
| Alphabetical listing | General | DBA-44 |
| Alphabetical listing | General | DDIT3 (pending) |
| Alphabetical listing | General | deltaNp63 |
| Alphabetical listing | General | desmin |
| Alphabetical listing | General | dicer |
| Alphabetical listing | General | DOG1 |
| Alphabetical listing | General | DPC4 / SMAD4 |
| Alphabetical listing | General | DUX4 |
| Alphabetical listing | General | E-cadherin |
| Alphabetical listing | General | EBER1 / 2 / LMP |
| Alphabetical listing | General | EBNA2 (pending) |
| Alphabetical listing | General | EGFR mutation specific antibodies |
| Alphabetical listing | General | elastic fibers |
| Alphabetical listing | General | EMA |
| Alphabetical listing | General | EpCAM |
| Alphabetical listing | General | ERG |
| Alphabetical listing | General | estrogen receptor (ER) |
| Alphabetical listing | General | ETV6-NTRK3 |
| Alphabetical listing | General | EWSR1 |
| Alphabetical listing | General | EZH2 |
| Alphabetical listing | General | Factor VIII |
| Alphabetical listing | General | Factor XIIIa |
| Alphabetical listing | General | Fas/CD95 |
| Alphabetical listing | General | Fas ligand/CD178 |
| Alphabetical listing | General | fascin |
| Alphabetical listing | General | filaggrin |
| Alphabetical listing | General | FLI1 |
| Alphabetical listing | General | FMC7 |
| Alphabetical listing | General | FN1 (pending) |
| Alphabetical listing | General | Fontana-Masson |
| Alphabetical listing | General | FOS (pending) |
| Alphabetical listing | General | FOSB (pending) |
| Alphabetical listing | General | FOXL2 |
| Alphabetical listing | General | FOXP3 |
| Alphabetical listing | General | fumarate hydratase |
| Alphabetical listing | General | Galectin3 |
| Alphabetical listing | General | gastrin (pending) |
| Alphabetical listing | General | GATA3 |
| Alphabetical listing | General | GCDFP-15 |
| Alphabetical listing | General | GCET (pending) |
| Alphabetical listing | General | GD2 (pending) |
| Alphabetical listing | General | GFAP |
| Alphabetical listing | General | Giemsa / Helicobacter pylori |
| Alphabetical listing | General | GLUT1 |
| Alphabetical listing | General | glutamine synthetase (pending) |
| Alphabetical listing | General | glycophorin A |
| Alphabetical listing | General | glypican 3 |
| Alphabetical listing | General | GMS |
| Alphabetical listing | General | gram stain |
| Alphabetical listing | General | granzyme B, M |
| Alphabetical listing | General | H3.3 G34W (H3F3A) |
| Alphabetical listing | General | H3K27M (pending) |
| Alphabetical listing | General | H3K27me3 |
| Alphabetical listing | General | H3K36M (pending) |
| Alphabetical listing | General | Hansel |
| Alphabetical listing | General | HBME |
| Alphabetical listing | General | hCG |
| Alphabetical listing | General | HepPar1 |
| Alphabetical listing | General | HER2 breast |
| Alphabetical listing | General | HER2 colon |
| Alphabetical listing | General | HER2 stomach GEJ |
| Alphabetical listing | General | HGAL |
| Alphabetical listing | General | HHF35 |
| Alphabetical listing | General | HHV8 |
| Alphabetical listing | General | HIK1083 |
| Alphabetical listing | General | HLA-DR |
| Alphabetical listing | General | HMB45 |
| Alphabetical listing | General | HNF1β (pending) |
| Alphabetical listing | General | HMGA2 |
| Alphabetical listing | General | HPL |
| Alphabetical listing | General | HPV |
| Alphabetical listing | General | hSNF5 |
| Alphabetical listing | General | HSP70 (pending) |
| Alphabetical listing | General | HSV (pending) |
| Alphabetical listing | General | IDH1 (R132H) |
| Alphabetical listing | General | IFITM1 |
| Alphabetical listing | General | IgA IgD IgG IgM (pending) |
| Alphabetical listing | General | IgG4 |
| Alphabetical listing | General | IMP3 |
| Alphabetical listing | General | inhibin |
| Alphabetical listing | General | INI1 |
| Alphabetical listing | General | INSM1 |
| Alphabetical listing | General | IRF4 |
| Alphabetical listing | General | iron |
| Alphabetical listing | General | islet 1 (Isl1) (pending) |
| Alphabetical listing | General | Jones methenamine silver (JMS) |
| Alphabetical listing | General | kappa & lambda light chains |
| Alphabetical listing | General | Ki67 |
| Alphabetical listing | General | KSHV |
| Alphabetical listing | General | L1-CAM (pending) |
| Alphabetical listing | General | LANA |
| Alphabetical listing | General | Leder |
| Alphabetical listing | General | LEF1 |
| Alphabetical listing | General | Leu7 |
| Alphabetical listing | General | LFABP (pending) |
| Alphabetical listing | General | lipid |
| Alphabetical listing | General | lipochrome |
| Alphabetical listing | General | LMO2 |
| Alphabetical listing | General | Luxol fast blue |
| Alphabetical listing | General | lysozyme |
| Alphabetical listing | General | mammaglobin |
| Alphabetical listing | General | mast cell tryptase (MCT) (pending) |
| Alphabetical listing | General | MART1 |
| Alphabetical listing | General | MCPyV |
| Alphabetical listing | General | MDM2 |
| Alphabetical listing | General | MDR |
| Alphabetical listing | General | MDR3 (pending) |
| Alphabetical listing | General | MEF2B |
| Alphabetical listing | General | MelanA |
| Alphabetical listing | General | MIB1 |
| Alphabetical listing | General | mic2 |
| Alphabetical listing | General | microphthalmia transcription factor (MITF) |
| Alphabetical listing | General | MLH1 |
| Alphabetical listing | General | MLH3 (pending) |
| Alphabetical listing | General | MOC31 |
| Alphabetical listing | General | Movat pentachrome (pending) |
| Alphabetical listing | General | MSA |
| Alphabetical listing | General | MSH1 |
| Alphabetical listing | General | MSH2 |
| Alphabetical listing | General | MSH6 |
| Alphabetical listing | General | MSI-Lynch syndrome |
| Alphabetical listing | General | mucins |
| Alphabetical listing | General | MUC1 |
| Alphabetical listing | General | MUC2-6 |
| Alphabetical listing | General | MUM1 |
| Alphabetical listing | General | MYB |
| Alphabetical listing | General | MYC |
| Alphabetical listing | General | myeloperoxidase |
| Alphabetical listing | General | MyoD1 |
| Alphabetical listing | General | myogenin / myoglobin |
| Alphabetical listing | General | NB84 (pending) |
| Alphabetical listing | General | NADH |
| Alphabetical listing | General | N-CAM |
| Alphabetical listing | General | NCCT (pending) |
| Alphabetical listing | General | Napsin A |
| Alphabetical listing | General | NeuN |
| Alphabetical listing | General | neuron specific enolase |
| Alphabetical listing | General | NF1 / NF2 |
| Alphabetical listing | General | NFP / neurofilament |
| Alphabetical listing | General | NGFR / p75 (pending) |
| Alphabetical listing | General | NKI-C3 |
| Alphabetical listing | General | NKX2 (pending) |
| Alphabetical listing | General | NKX3.1 |
| Alphabetical listing | General | nonspecific esterase |
| Alphabetical listing | General | NPM1 |
| Alphabetical listing | General | NTRK |
| Alphabetical listing | General | NUTM1 |
| Alphabetical listing | General | OCT2 |
| Alphabetical listing | General | OCT3/4 |
| Alphabetical listing | General | Oil Red O |
| Alphabetical listing | General | Olig2 |
| Alphabetical listing | General | OTP (orthopedia) (pending) |
| Alphabetical listing | General | P glycoprotein |
| Alphabetical listing | General | p16 |
| Alphabetical listing | General | p21 |
| Alphabetical listing | General | p40 |
| Alphabetical listing | General | p53 |
| Alphabetical listing | General | p57 |
| Alphabetical listing | General | p62 (pending) |
| Alphabetical listing | General | p63 |
| Alphabetical listing | General | p120 catenin |
| Alphabetical listing | General | P504S |
| Alphabetical listing | General | Pan-TRK |
| Alphabetical listing | General | PAP |
| Alphabetical listing | General | PAS |
| Alphabetical listing | General | PAX2 |
| Alphabetical listing | General | PAX5 |
| Alphabetical listing | General | PAX8 |
| Alphabetical listing | General | PCNA |
| Alphabetical listing | General | PD-1 |
| Alphabetical listing | General | PDL1 22C3 |
| Alphabetical listing | General | PDL1 SP142 |
| Alphabetical listing | General | PDL1 SP263 (pending) |
| Alphabetical listing | General | PECAM1 |
| Alphabetical listing | General | perforin (pending) |
| Alphabetical listing | General | PGP9.5 (pending) |
| Alphabetical listing | General | PHLDA1 (pending) |
| Alphabetical listing | General | phosphohistone H3 |
| Alphabetical listing | General | Pit1 |
| Alphabetical listing | General | PLAG1 |
| Alphabetical listing | General | PLAP |
| Alphabetical listing | General | PMS2 |
| Alphabetical listing | General | podoplanin |
| Alphabetical listing | General | PRAME |
| Alphabetical listing | General | prealbumin (pending) |
| Alphabetical listing | General | progesterone receptor (PR) |
| Alphabetical listing | General | prostein |
| Alphabetical listing | General | PROX1 (pending) |
| Alphabetical listing | General | PSA |
| Alphabetical listing | General | PSAP |
| Alphabetical listing | General | PSMA |
| Alphabetical listing | General | PTEN |
| Alphabetical listing | General | PU.1 |
| Alphabetical listing | General | RB1 |
| Alphabetical listing | General | RCC |
| Alphabetical listing | General | reticulin |
| Alphabetical listing | General | ROS1 |
| Alphabetical listing | General | S100 |
| Alphabetical listing | General | S100P |
| Alphabetical listing | General | SALL4 |
| Alphabetical listing | General | SATB2 |
| Alphabetical listing | General | SDH (pending) |
| Alphabetical listing | General | SDHB |
| Alphabetical listing | General | serum amyloid A/SAA |
| Alphabetical listing | General | SF1 |
| Alphabetical listing | General | Sirius red |
| Alphabetical listing | General | SMAD4 / DPC4 |
| Alphabetical listing | General | SMARCA4 |
| Alphabetical listing | General | SMMHC / smooth muscle myosin heavy chain |
| Alphabetical listing | General | smoothelin |
| Alphabetical listing | General | SOX2 |
| Alphabetical listing | General | SOX9 |
| Alphabetical listing | General | SOX10 |
| Alphabetical listing | General | SOX11 |
| Alphabetical listing | General | SOX17 |
| Alphabetical listing | General | SSTR2A |
| Alphabetical listing | General | STAT6 |
| Alphabetical listing | General | Sudan Black B |
| Alphabetical listing | General | SV40 (pending) |
| Alphabetical listing | General | synaptophysin |
| Alphabetical listing | General | SYT / SSX (pending) |
| Alphabetical listing | General | TCL1 |
| Alphabetical listing | General | TCR (pending) |
| Alphabetical listing | General | TDP-43 (pending) |
| Alphabetical listing | General | TdT |
| Alphabetical listing | General | TERT |
| Alphabetical listing | General | TFE3 |
| Alphabetical listing | General | thrombomodulin |
| Alphabetical listing | General | thyroglobulin |
| Alphabetical listing | General | TLE1 |
| Alphabetical listing | General | Tpit |
| Alphabetical listing | General | TRAP |
| Alphabetical listing | General | treponema IHC |
| Alphabetical listing | General | trichrome |
| Alphabetical listing | General | TROP2 (pending) |
| Alphabetical listing | General | trypsin (pending) |
| Alphabetical listing | General | TSC1/2 |
| Alphabetical listing | General | TTF1 |
| Alphabetical listing | General | TSH (pending) |
| Alphabetical listing | General | tyrosinase |
| Alphabetical listing | General | Unknown primary |
| Alphabetical listing | General | uric acid |
| Alphabetical listing | General | uroplakin II |
| Alphabetical listing | General | uroplakin III |
| Alphabetical listing | General | VHL |
| Alphabetical listing | General | villin |
| Alphabetical listing | General | vimentin |
| Alphabetical listing | General | vWF |
| Alphabetical listing | General | Warthin-Starry |
| Alphabetical listing | General | Wright-Giemsa |
| Alphabetical listing | General | WT1 |
| Alphabetical listing | General | YAP1 |
| Alphabetical listing | General | Ziehl-Neelsen |
| Alphabetical listing | Superpages | entire chapter |
| Alphabetical listing | Superpages | images |
| Alphabetical listing | Superpages | virtual slides |
Inmunotinciones y Tinciones Especiales Comunes en Hematopatología
| Marcador | Células/Procesos Asociados |
|---|---|
CD1a |
Células de Langerhans, linfocitos T inmaduros |
CD2 |
Linfocitos T/NK; mastocitos atípicos |
CD3 |
Linfocitos T |
CD4 |
Linfocitos T cooperadores; monocitos/macrófagos |
CD5 |
Linfocitos T |
CD7 |
Linfocitos T/NK |
CD8 |
Linfocitos T citotóxicos |
CD10 |
Hematogones/Linfoblastos B; centro germinal de linfocitos B; granulocitos maduros |
CD11b |
Monocitos, neutrófilos, linfocitos NK |
CD11c |
Leucemia de células peludas |
CD13 |
Granulocitos/Monocitos |
CD14 |
Monocitos maduros |
CD15 |
Granulocitos (de mielocito a neutrófilo); monocitos; células de Reed-Sternberg |
CD19 |
Linfocitos B inmaduros y maduros; células plasmáticas |
CD20 |
Linfocitos B maduros |
CD21 |
Células dendríticas |
CD22 |
Leucemia de células peludas |
CD23 |
Células dendríticas, linfocitos B reactivos |
CD25 |
Leucemia de células peludas; mastocitos atípicos; leucemia/linfoma de linfocitos T del adulto |
CD27 |
Células plasmáticas normales |
CD30 |
Inmunoblastos; células de Reed-Sternberg; algunos linfomas de linfocitos B grandes; linfoma anaplásico de células grandes (ALK+/-); linfoma de linfocitos T de transformación de células grandes |
CD33 |
Granulocitos/monocitos |
CD34 |
Células inmaduras |
CD36 |
Monocitos |
CD42 |
Megacariocitos |
CD45 |
Células hematolinfoides (puede ser negativo en leucemia/linfoma linfoblástico y mieloma múltiple/células plasmáticas) |
CD56 |
Células NK; células de mieloma de células plasmáticas |
CD61 |
Megacariocitos |
CD68 |
Histiocitos |
CD71 |
Precursores eritroides |
CD79a |
Linfocitos B inmaduros y maduros; células plasmáticas |
CD81 |
Linfocitos B inmaduros; células plasmáticas |
CD117 |
Precursores eritroides/granulocíticos; mastocitos |
CD138 |
Células plasmáticas |
CD163 |
Histiocitos |
Inmunotinciones y Tinciones Especiales Comunes en Patología (con enfoque en Dermatopatología)
| Tinción | Comúnmente positivo en... |
|---|---|
| Tinciones Inmunohistoquímicas | |
CD1a |
Células de Langerhans (e histiocitosis de Langerhans) |
CD3 |
Todos los linfocitos T normales |
CD4 |
Linfocitos T cooperadores, histiocitos/macrófagos, células de Langerhans |
CD5 |
Todos los linfocitos T normales (y algunos linfomas de linfocitos B como LLC y linfoma de células del manto) |
CD7 |
Todos los linfocitos T normales |
CD8 |
Linfocitos T citotóxicos |
CD20 |
Linfocitos B (y la mayoría de los linfomas de linfocitos B) |
CD45 |
Glóbulos blancos normales. La mayoría de los linfomas y leucemias. No en mieloma. |
CD31 |
Células endoteliales (vasos sanguíneos y linfáticos) |
CD34 |
Células endoteliales (vasos sanguíneos y +/- linfáticos). DFSP. Muchos otros. |
ERG
|
Células endoteliales (vasos sanguíneos y linfáticos). LMA. Carcinoma de próstata. Otros. |
CD68 & CD163 |
Histiocitos/macrófagos |
CD99 |
Sarcoma de Ewing. La mayoría de los tumores de células azules redondas. Muchos otros. |
CD117 (ckit) |
Mastocitos, incluyendo mastocitosis. Muchos otros. |
| Triptasa de Mastocitos | Mastocitos, incluyendo mastocitosis. |
Citoqueratinas (CK) (existen muchas queratinas; a continuación se enumeran algunas...) |
Todas las células epiteliales (normales o neoplásicas) en cualquier parte del cuerpo (p. ej., epidermis, glándulas sudoríparas, folículos pilosos, todos los carcinomas [raras excepciones]). |
CK7 |
Epitelio normal de mama y pulmón (y adenocarcinoma de mama/pulmón). Carcinoma de células pequeñas de pulmón. Otros. |
CK20 |
Epitelio normal de colon. Adenocarcinomas colorrectales. Carcinoma de células de Merkel ("puntiforme"). Otros. |
CK5/6 |
Epidermis normal. CEC. CBC. Otros. |
D240 (podoplanina) |
Células endoteliales linfáticas |
| Desmina | Músculo liso, músculo esquelético, neoplasias musculares (leio- y rabdo- tumores) |
| Actina de músculo liso | Músculo liso, miofibroblastos y algunas otras cosas |
Factor XIIIa |
Dermatofibroma. Negativo en DFSP. |
VHH-8 |
Sarcoma de Kaposi (y algunas otras causas oscuras causadas por VHH8) |
HMB-45, MART-1/Melan-A, MiTF |
Melanocitos normales, nevos, melanoma |
S100 |
Melanocitos normales, nevos, melanoma. Células de Langerhans. Nervios (células de Schwann). Muchas otras cosas. Marcador MUY sensible para tumores melanocíticos, pero no específico. |
SOX-10 |
Melanocitos normales, nevos, melanoma. Nervios (células de Schwann). Similar a S100 pero un poco más específico (y más bonito/menos tinción de fondo, también) |
p63 & p40 |
Epidermis, glándulas sudoríparas, folículos pilosos, CEC, CBC, la mayoría de los tumores anexiales de la piel. Muchos otros. Generalmente negativo en adenocarcinomas de órganos viscerales. |
| Tinciones Histoquímicas ("especiales") | |
AFB (Fite, Kinyoun y Ziehl-Neelsen son tipos de tinción AFB) |
Tiñe micobacterias de rojo (Fite es mejor para M. leprae, por lo que típicamente usamos Fite en dermatopatología) |
GMS |
Tiñe levaduras y hifas fúngicas de negro |
PAS con diastasa (PASD) |
Tiñe levaduras y hifas fúngicas de rosa/rojo |
| Giemsa | Tiñe mastocitos de morado |
| Tricrómico | Tiñe el músculo de rojo brillante, tiñe el colágeno de azul oscuro (o verde). |
Verhoeff-Van Gieson (VVG) |
Tiñe las fibras elásticas de negro |
| Rojo Congo | Tiñe amiloide de rojo/"salmón" (luz normal) y "verde manzana" (luz polarizada) |
| Tioflavina T | Tiñe amiloide de verde brillante (se debe observar con microscopio de inmunofluorescencia) |